

Secuenciación del genoma de novo de plantas/animales




Ventajas del servicio
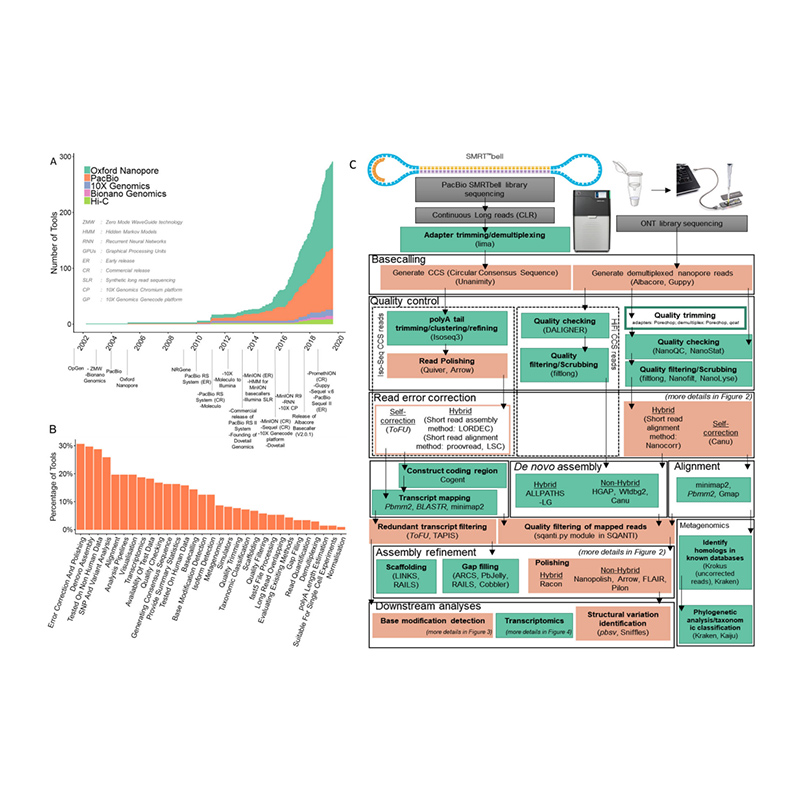
Desarrollo de plataformas de secuenciación y bioinformática ende novoensamblaje del genoma
(Amarasinghe SL et al.,Biología del genoma, 2020)
● Construir genomas novedosos y mejorar los genomas de referencia existentes para especies de interés.
● Mayor precisión, continuidad e integridad en el montaje.
● Construcción de recursos fundamentales para la investigación en polimorfismo de secuencia, QTL, edición de genes, mejoramiento, etc.
● Equipado con un espectro completo de plataformas de secuenciación de tercera generación: solución integral de ensamblaje del genoma
● Estrategias flexibles de secuenciación y ensamblaje que satisfacen diversos genomas con diferentes características
● Equipo bioinformático altamente cualificado y con gran experiencia en ensamblajes de genomas complejos, incluyendo poliploides, genomas gigantes, etc.
● Más de 100 casos exitosos con un factor de impacto publicado acumulativo de más de 900
● El tiempo de respuesta es tan rápido como 3 meses para el ensamblaje del genoma a nivel de cromosoma.
● Sólido soporte técnico con una serie de patentes y derechos de autor de software tanto en la vertiente experimental como en bioinformática.
Especificaciones de servicio
|
Contenido
|
Plataforma
|
Longitud de lectura
|
Cobertura
|
| Encuesta del genoma
| Illumina NovaSeq
| PE150
| ≥50X
|
| Secuenciación del genoma
| PacBio Revio
| Lecturas de alta fidelidad de 15 kb
| ≥30X
|
| Hola-C
| Illumina NovaSeq
| PE150
| ≥100X
|
flujo de trabajo

Requisitos de muestra y entrega
Requisitos de muestra:
| Especies | Tejido | Para PacBio | Para nanoporos |
| animales | Órganos viscerales (hígado, bazo, etc.) | ≥ 1,0 gramos | ≥ 3,5 gramos |
| Músculo | ≥ 1,5 gramos | ≥ 5,0 gramos | |
| Sangre de mamíferos | ≥ 1,5 ml | ≥ 5,0 ml | |
| Sangre de peces o pájaros. | ≥ 0,2 ml | ≥ 0,5 ml | |
| Plantas | hojas frescas | ≥ 1,5 gramos | ≥ 5,0 gramos |
| Pétalo o tallo | ≥ 3,5 gramos | ≥ 10,0 gramos | |
| Raíces o semillas | ≥ 7,0 gramos | ≥ 20,0 gramos | |
| Células | Cultivo de células | ≥3×107 | ≥1×108 |
Entrega de muestra recomendada
Envase: tubo de centrífuga de 2 ml (no se recomienda papel de aluminio)
Para la mayoría de las muestras, recomendamos no conservarlas en etanol.
Etiquetado de muestras: las muestras deben estar claramente etiquetadas y ser idénticas al formulario de información de muestra enviado.
Envío: Hielo seco: las muestras deben empaquetarse primero en bolsas y enterrarse en hielo seco.
Flujo de trabajo del servicio

Diseño de experimentos

Entrega de muestra

Extracción de ADN

construcción de biblioteca

Secuenciación

Análisis de los datos

Servicios postventa
*Los resultados de demostración que se muestran aquí provienen todos de genomas publicados con Biomarker Technologies
1.Circos sobre el ensamblaje del genoma a nivel cromosómico deG. rotundifoliumpor plataforma de secuenciación Nanopore
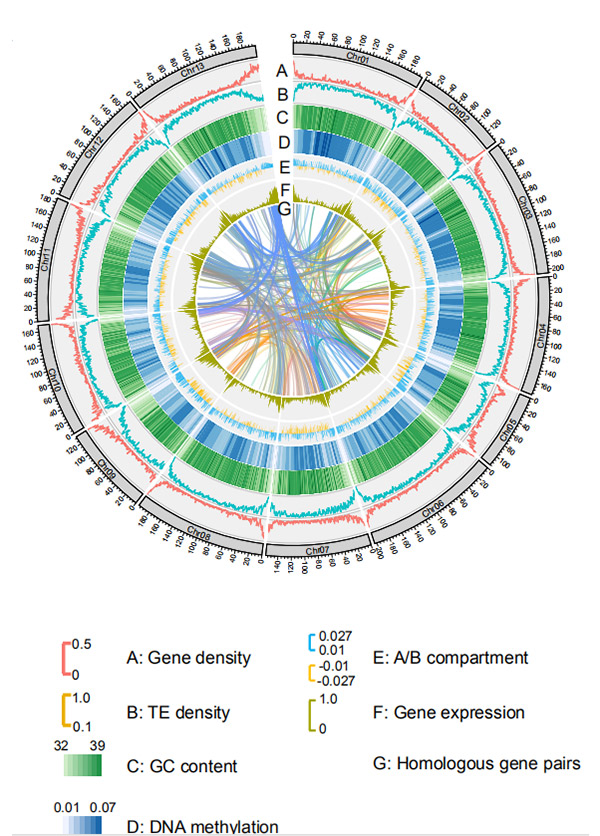
Wang M et al.,Biología molecular y evolución, 2021
2.Estadísticas del ensamblaje y anotación del genoma del centeno Weining
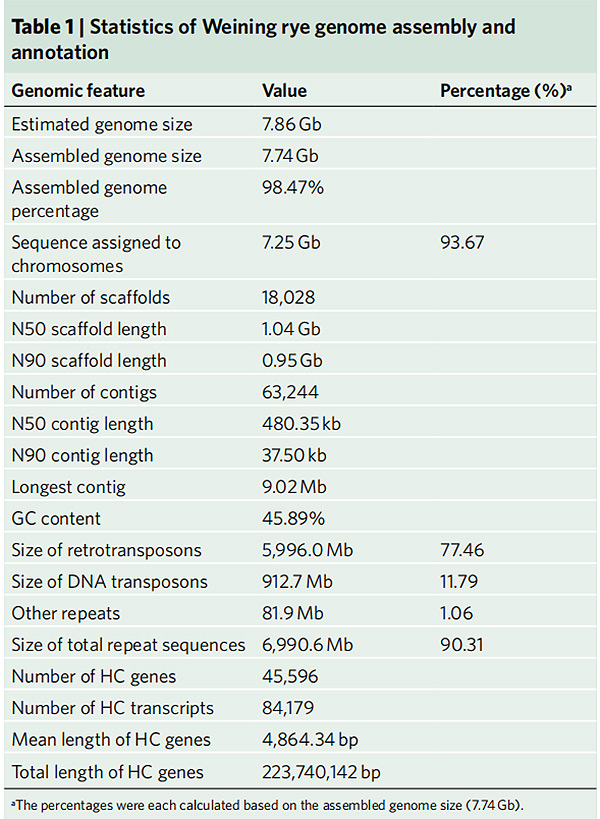
Li G et al.,Genética de la naturaleza, 2021
3.Predicción genética deSechium edulegenoma, derivado de tres métodos de predicción:De novopredicción, predicción basada en homología y predicción basada en datos de RNA-Seq
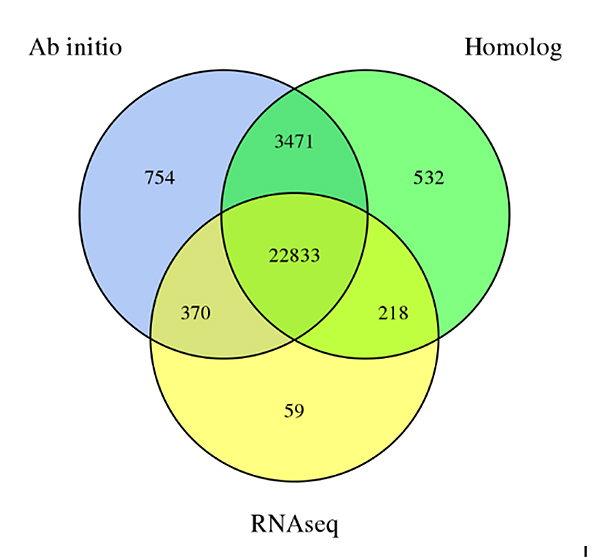
Fu A y otros,Investigación en horticultura, 2021
4.Identificación de repeticiones terminales largas intactas en tres genomas de algodón.
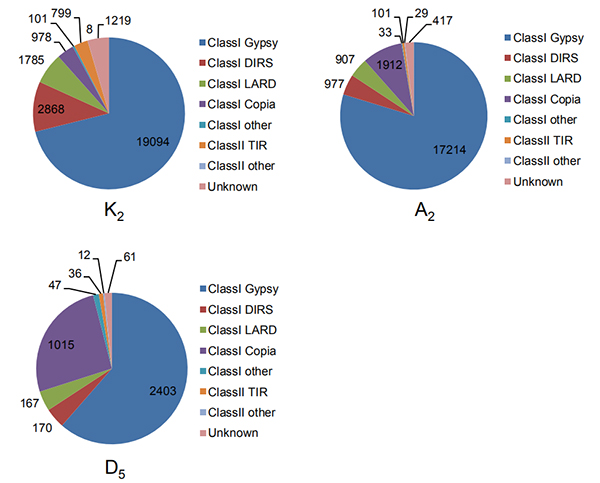
Wang M et al.,Biología molecular y evolución, 2021
5.Mapa de calor Hi-C delC. acuminatagenoma que muestra interacciones generales en todo el genoma.La intensidad de las interacciones Hi-C es proporcional a la distancia lineal entre contigs.Una línea recta limpia en este mapa de calor indica un anclaje muy preciso de los cóntigos en los cromosomas.(Relación de anclaje Contig: 96,03%)
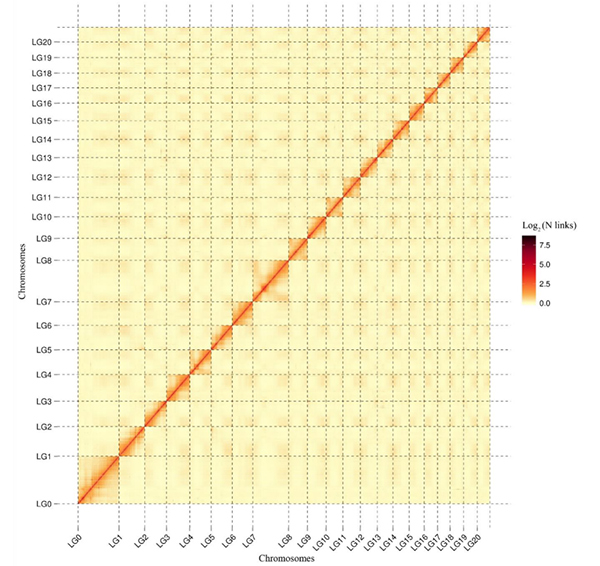
Kang M et al.,comunicaciones de la naturaleza,2021
Caso BMK
Un ensamblaje genómico de alta calidad destaca las características genómicas del centeno y los genes agronómicamente importantes
Publicado: Genética de la naturaleza, 2021
Estrategia de secuenciación:
Ensamblaje del genoma: modo PacBio CLR con biblioteca de 20 kb (497 Gb, aprox. 63×)
Corrección de secuencia: NGS con biblioteca de ADN de 270 pb (430 Gb, aprox. 54×) en plataforma Illumina
Anclaje de Contigs: biblioteca Hi-C (560 Gb, aprox. 71×) en plataforma Illumina
Mapa óptico: (779,55 Gb, aprox. 99×) en Bionano Irys
Resultados clave
1. Se publicó un ensamblaje del genoma del centeno Weining con un tamaño total del genoma de 7,74 Gb (98,74% del tamaño del genoma estimado mediante citometría de flujo).El andamio N50 de este conjunto alcanzó 1,04 Gb.El 93,67% de los cóntigos se anclaron con éxito en 7 pseudocromosomas.Esta asamblea fue evaluada por mapa de vinculación, LAI y BUSCO, lo que resultó en puntajes altos en todas las evaluaciones.
2. Se realizaron más estudios sobre genómica comparada, mapa de ligamiento genético y estudios de transcriptómica sobre la base de este genoma.Se revelaron una serie de rasgos genómicos relacionados, incluidas las duplicaciones de genes en todo el genoma y su impacto en los genes de biosíntesis de almidón;organización física de loci complejos de prolamina, características de expresión genética subyacentes al rasgo de encabezado temprano y regiones cromosómicas y loci putativos asociados a la domesticación en el centeno.
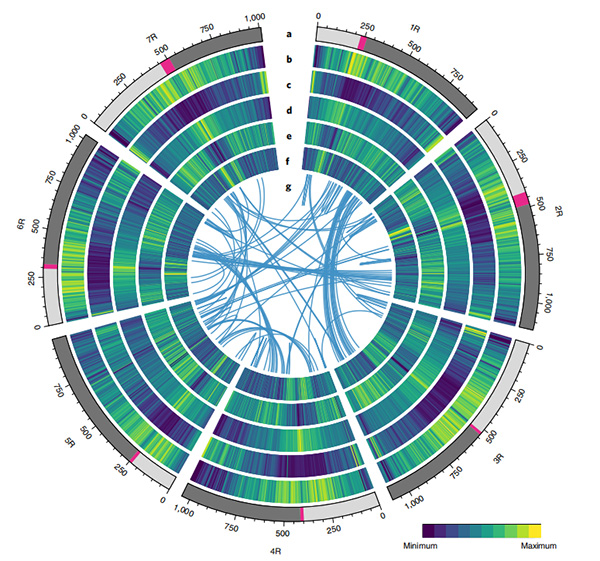 Diagrama circos sobre las características genómicas del genoma del centeno Weining | 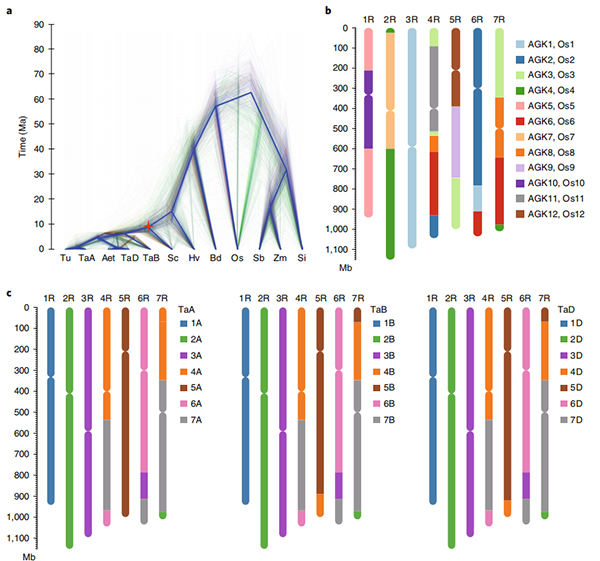 Análisis evolutivos y de sintenia cromosómica del genoma del centeno. |
Li, G., Wang, L., Yang, J.et al.Un ensamblaje genómico de alta calidad resalta las características genómicas del centeno y los genes agronómicamente importantes.Nat Genet 53,574–584 (2021).
https://doi.org/10.1038/s41588-021-00808-z





