

Bulked Segregant -analyysi






Palvelun edut
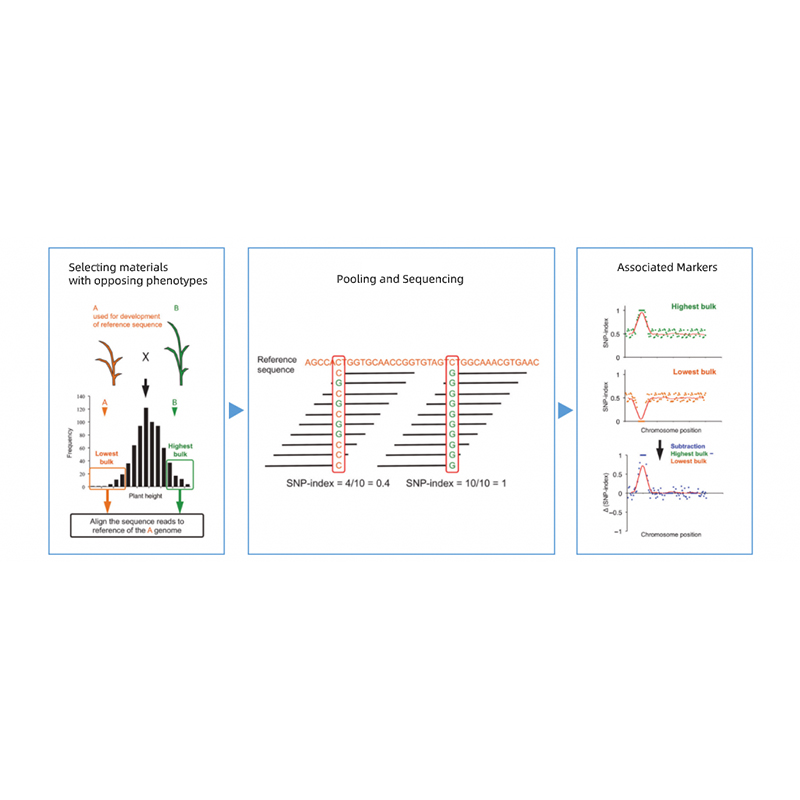
Takagi et al., The Plant Journal, 2013
● Tarkka paikannus: bulkkien sekoittaminen 30+30 - 200+200 yksilöllä taustamelun minimoimiseksi;ei-synonyymeihin mutantteihin perustuva ehdokasalueen ennuste.
● Kattava analyysi: syvällinen ehdokasgeenifunktion annotaatio, mukaan lukien NR, SwissProt, GO, KEGG, COG, KOG jne.
● Nopeampi läpimenoaika: Nopea geenin lokalisointi 45 työpäivän sisällä.
● Laaja kokemus: BMK on osallistunut tuhansien piirteiden lokalisointiin, kattaen erilaisia lajeja, kuten kasveja, vesituotteita, metsää, kukkia, hedelmiä jne.
Palvelun tiedot
Väestö:
Vastakkaisten fenotyyppien vanhempien jälkeläisten erottelu.
esim. F2-jälkeläiset, Backcrossing (BC), Rekombinantti sisäsiittolinja (RIL)
Sekoitusallas
Laadulliset ominaisuudet: 30-50 yksilöä (vähintään 20) / irtotavara
Kvantitatiiviset piirteet: 5–10 % yksilöitä, joilla on jompikumpi äärimmäisistä fenotyypeistä koko populaatiosta (vähintään 30+30).
Suositeltu sekvensointisyvyys
Vähintään 20X/vanhempi ja 1X/jälkeläisyksityiskohta (esim. 30+30 yksilön jälkeläisten sekoituspoolille, sekvensointisyvyys on 30X/bulkki)
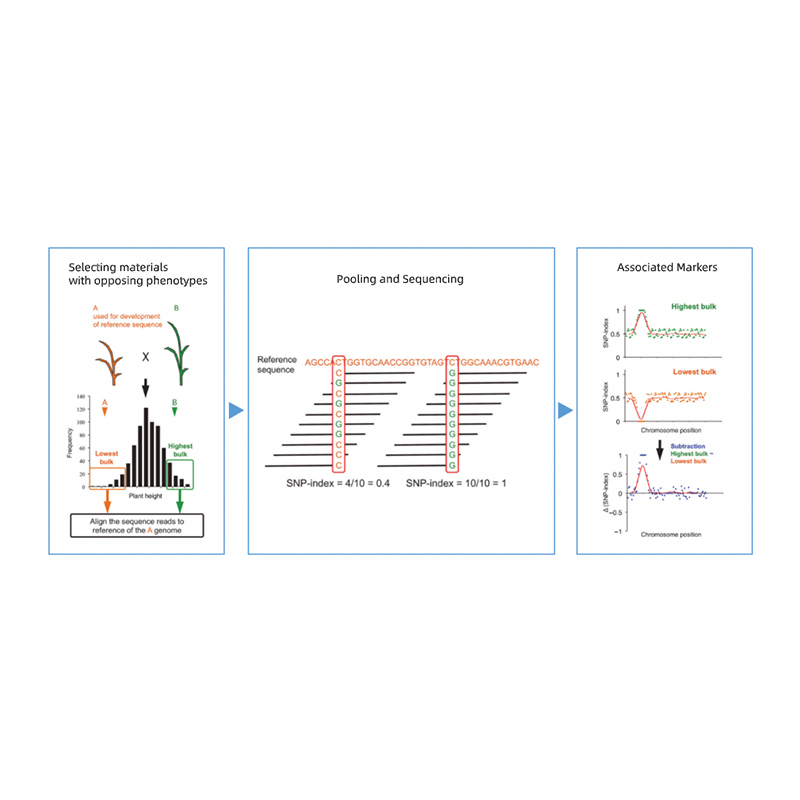
Bioinformatiikan analyysit
● Koko genomin uudelleensekvensointi
● Tietojen käsittely
● SNP/Indel-puhelut
● Ehdokasalueen seulonta
● Ehdokasgeenifunktion huomautus
Näytevaatimukset ja toimitus
Esimerkkivaatimukset:
Nukleotidit:
| gDNA-näyte | Kudosnäyte |
| Pitoisuus: ≥30 ng/μl | Kasvit: 1-2 g |
| Määrä: ≥2 μg (tilavuus ≥15 μl) | Eläimet: 0,5-1 g |
| Puhtaus: OD260/280 = 1,6-2,5 | Kokoveri: 1,5 ml |
Palvelun työnkulku

Kokeilusuunnittelu

Näytteen toimitus

RNA:n uuttaminen

Kirjaston rakentaminen

Jaksotus

Tietojen analysointi

Myynnin jälkeiset palvelut
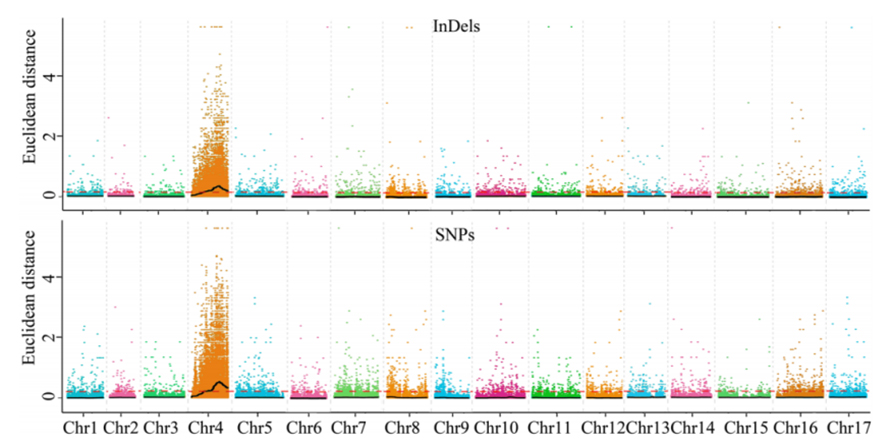
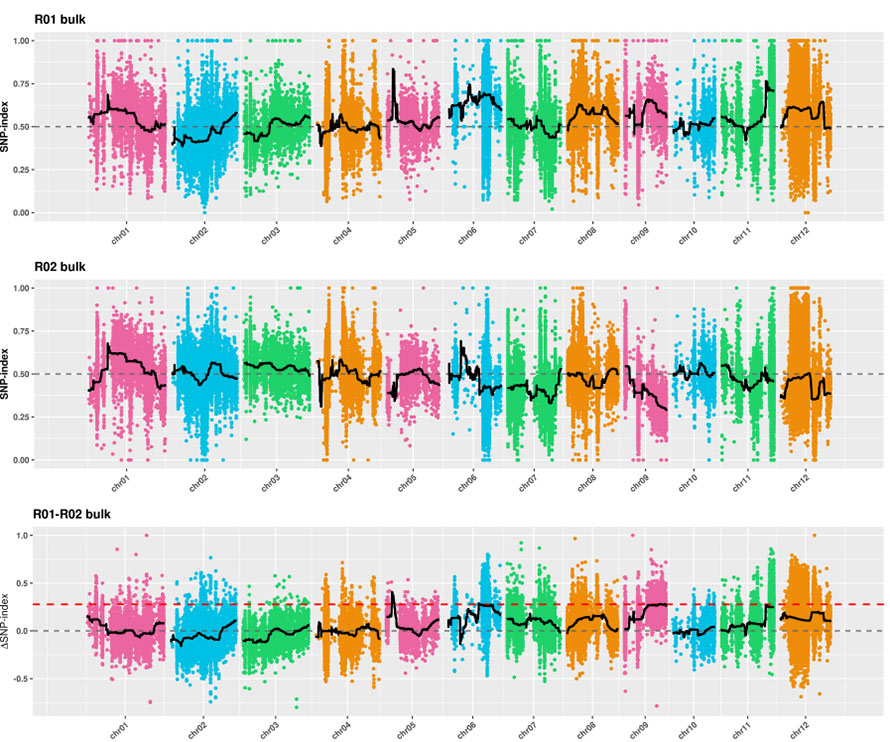
1. Assosiaatioanalyysi perustuu euklidiseen etäisyyteen (ED) ehdokasalueen tunnistamiseksi.Seuraavassa kuvassa
X-akseli: kromosomiluku;Jokainen piste edustaa SNP:n ED-arvoa.Musta viiva vastaa sovitettua ED-arvoa.Korkeampi ED-arvo osoittaa merkittävämmän yhteyden paikan ja fenotyypin välillä.Punainen katkoviiva edustaa merkittävän assosioinnin kynnystä.
2. Assosiaatioanalyysi ei perustu SNP-indeksiin
X-akseli: kromosomiluku;Jokainen piste edustaa SNP-indeksin arvoa.Musta viiva tarkoittaa sovitettua SNP-indeksin arvoa.Mitä suurempi arvo on, sitä merkittävämpi yhteys on.
BMK kotelo
Päävaikutukseltaan kvantitatiivisen ominaisuuden lokus Fnl7.1 koodaa myöhäisen alkion syntyvaiheen runsasta proteiinia, joka liittyy kurkun hedelmän kaulan pituuteen
Julkaistu: Plant Biotechnology Journal, 2020
Sekvensointistrategia:
Vanhemmat (Jin5-508, YN): Koko genomin uudelleensekvensointi 34× ja 20×.
DNA-poolit (50 pitkäkaulaista ja 50 lyhytkaulaista): Uudelleensekvensointi 61× ja 52×
Tärkeimmät tulokset
Tässä tutkimuksessa erottuva populaatio (F2 ja F2:3) luotiin risteyttämällä pitkäkaulainen kurkkulinja Jin5-508 ja lyhytkaulainen YN.Kaksi DNA-poolia rakennettiin 50 äärimmäisen pitkäkaulaisesta ja 50 äärimmäisen lyhytkaulaisesta yksilöstä.Merkittävä vaikutus QTL tunnistettiin Chr07:lle BSA-analyysillä ja perinteisellä QTL-kartoituksella.Ehdokasaluetta kavennettiin edelleen hienokartoituksella, geeniekspression kvantitatiivisella määrityksellä ja siirtogeenisillä kokeilla, jotka paljastivat avaingeenin kaulan pituuden säätelyssä, CsFnl7.1.Lisäksi polymorfismin CsFnl7.1-promoottorialueella havaittiin liittyvän vastaavaan ilmentymiseen.Lisäfylogeneettinen analyysi osoitti, että Fnl7.1-lokus on hyvin todennäköisesti peräisin Intiasta.
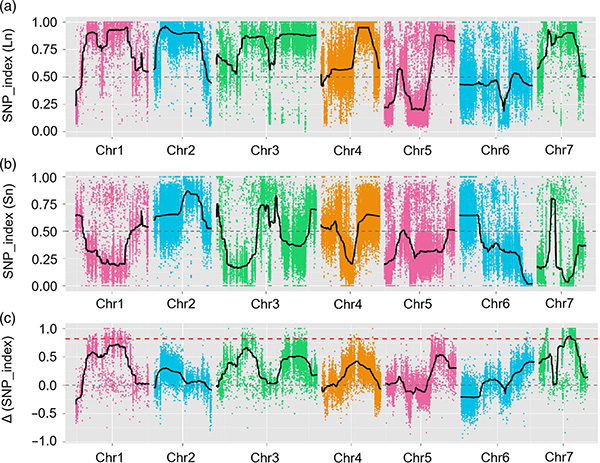 QTL-kartoitus BSA-analyysissä kurkun kaulan pituuteen liittyvän ehdokasalueen tunnistamiseksi | 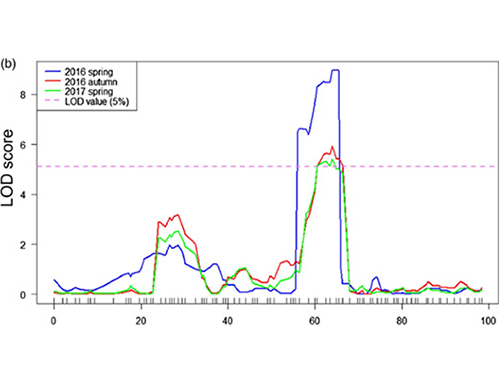 Chr07:ssä tunnistetun kurkun kaulan pituisen QTL:n LOD-profiilit |
Xu, X. et ai."Päävaikutteinen kvantitatiivinen ominaisuuslokus Fnl7.1 koodaa myöhäisen alkion syntyvaiheen runsasta proteiinia, joka liittyy kurkun hedelmän kaulan pituuteen."Plant Biotechnology Journal 18.7 (2020).





