

Kasvien/eläinten de Novo genomin sekvensointi




Palvelun edut
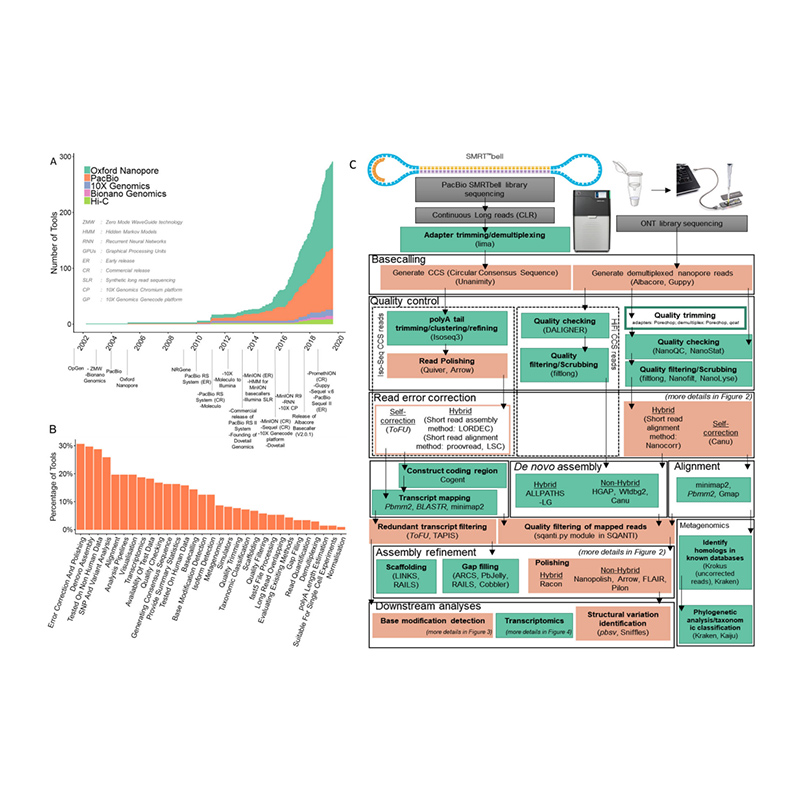
Sekvensointialustojen ja bioinformatiikan kehittäminende novogenomin kokoonpano
(Amarasinghe SL et ai.,Genomibiologia, 2020)
● Uusien genomien rakentaminen ja olemassa olevien vertailugenomien parantaminen kiinnostaville lajeille.
● Parempi kokoonpanon tarkkuus, jatkuvuus ja täydellisyys
● Perusresurssien rakentaminen sekvenssipolymorfismin, QTL:ien, geenien muokkaamisen, jalostuksen jne. tutkimukseen.
● Varustettu täydellä kirjolla kolmannen sukupolven sekvensointialustoja: yhden luukun genomin kokoonpanoratkaisu
● Joustavat sekvensointi- ja kokoamisstrategiat, jotka täyttävät erilaisia genomeja erilaisilla ominaisuuksilla
● Korkeasti koulutettu bioinformaatikkotiimi, jolla on suuri kokemus monimutkaisista genomikokoonpanoista, kuten polyploideista, jättiläisgenomeista jne.
● Yli 100 onnistunutta tapausta, joiden kumulatiivinen julkaistu vaikutuskerroin on yli 900
● Kromosomitason genomin kokoamisen läpimenoaika jopa 3 kuukaudessa.
● Vankka tekninen tuki, jossa on useita patentteja ja ohjelmistojen tekijänoikeuksia sekä kokeellisessa että bioinformatiikan alalla.
Palvelun tiedot
|
Sisältö
|
Alusta
|
Lue pituus
|
Kattavuus
|
| Genomitutkimus
| Illumina NovaSeq
| PE150
| ≥ 50X
|
| Genomin sekvensointi
| PacBio Revio
| 15 kb HiFi-luku
| ≥ 30X
|
| Hei-C
| Illumina NovaSeq
| PE150
| ≥100X
|
Työnkulku

Näytevaatimukset ja toimitus
Esimerkkivaatimukset:
| Laji | Kudos | PacBiolle | Nanoporelle |
| Eläimet | Viskeraaliset elimet (maksa, perna jne.) | ≥ 1,0 g | ≥ 3,5 g |
| Lihas | ≥ 1,5 g | ≥ 5,0 g | |
| Nisäkkäiden veri | ≥ 1,5 ml | ≥ 5,0 ml | |
| Kalojen tai lintujen veri | ≥ 0,2 ml | ≥ 0,5 ml | |
| Kasveja | Tuoreet lehdet | ≥ 1,5 g | ≥ 5,0 g |
| Terälehti tai varsi | ≥ 3,5 g | ≥ 10,0 g | |
| Juuret tai siemenet | ≥ 7,0 g | ≥ 20,0 g | |
| Solut | Soluviljely | ≥ 3×107 | ≥ 1×108 |
Suositeltu näytetoimitus
Säiliö: 2 ml sentrifugiputki (tinafoliota ei suositella)
Useimpien näytteiden osalta suosittelemme, ettei niitä säilytetä etanolissa.
Näytteen merkintä: Näytteiden on oltava selkeästi merkittyjä ja identtisiä näytetietolomakkeen kanssa.
Toimitus: Kuivajää: Näytteet on pakattava ensin pusseihin ja haudattava kuivajäähän.
Palvelun työnkulku

Kokeilusuunnittelu

Näytteen toimitus

DNA:n uuttaminen

Kirjaston rakentaminen

Jaksotus

Tietojen analysointi

Myynnin jälkeiset palvelut
*Tässä näkyvät esittelytulokset ovat kaikki Biomarker Technologiesin julkaisemista genomeista
1.Circos on kromosomitason genomikokoonpanoG. rotundifoliumNanopore-sekvensointialustalta
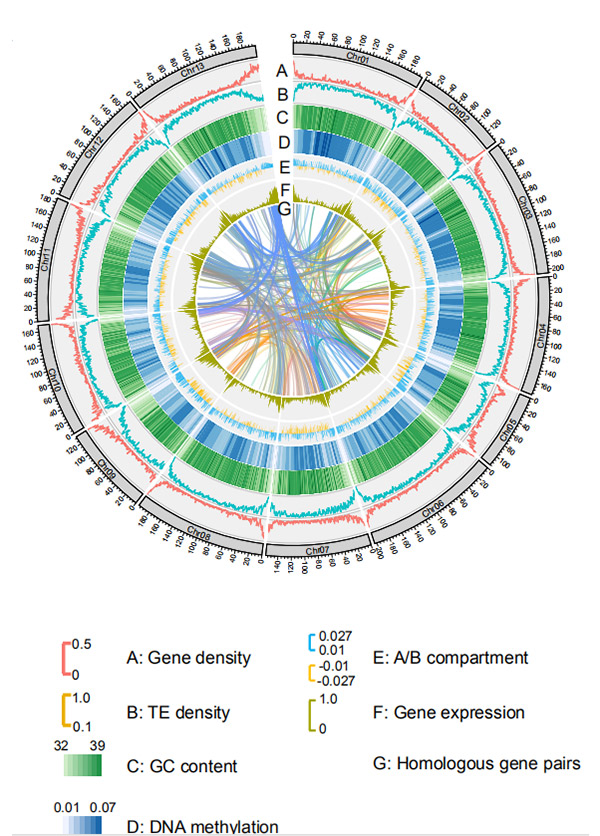
Wang M et ai.,Molekyylibiologia ja evoluutio, 2021
2. Weiningin rukiin genomin kokoonpanon ja annotoinnin tilastot
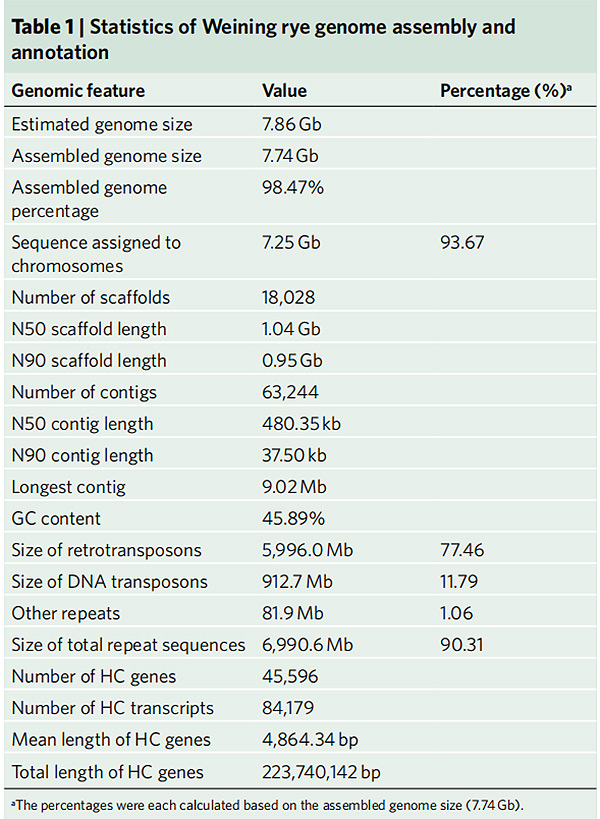
Li G et ai.,Luonnon genetiikka, 2021
3.Geenien ennusteSechium edulegenomi, joka on johdettu kolmesta ennustemenetelmästä:De novoennustus, homologiaan perustuva ennuste ja RNA-Seq-dataan perustuva ennuste
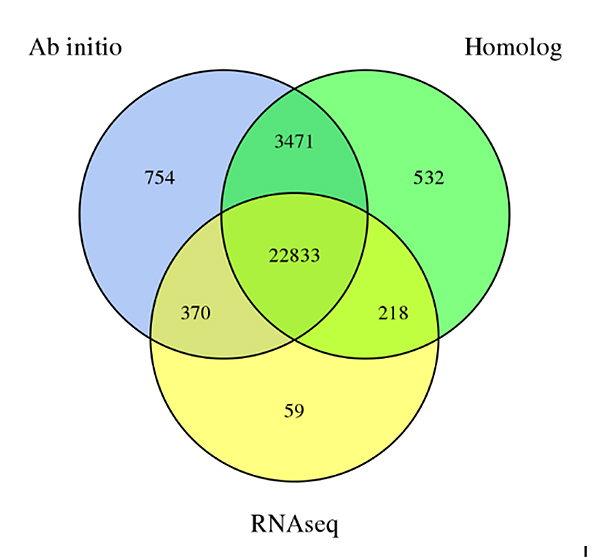
Fu A et ai.,Puutarhatutkimus, 2021
4. Intaktien pitkien terminaalisten toistojen tunnistaminen kolmessa puuvillan genomissa
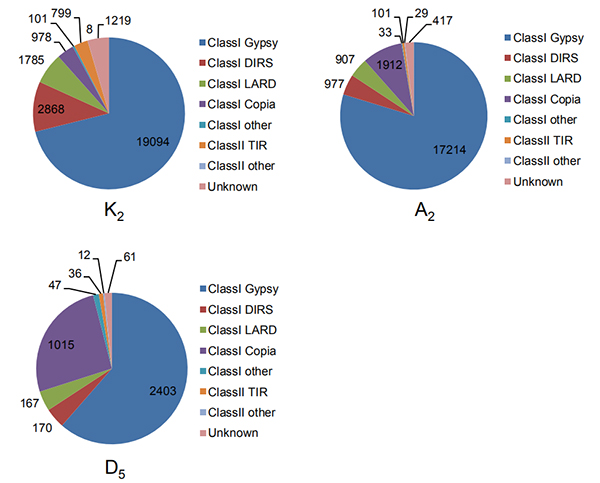
Wang M et ai.,Molekyylibiologia ja evoluutio, 2021
5.Hi-C lämpökarttaC. acuminatagenomi, joka osoittaa genominlaajuista all-by-all-vuorovaikutusta.Hi-C-vuorovaikutusten intensiteetti on verrannollinen jatkuvien kohtien väliseen lineaariseen etäisyyteen.Puhdas suora viiva tässä lämpökartassa osoittaa erittäin tarkan jatkuvien ankkuroitumisen kromosomeihin.(Jatkuvien ankkurointisuhde: 96,03 %)
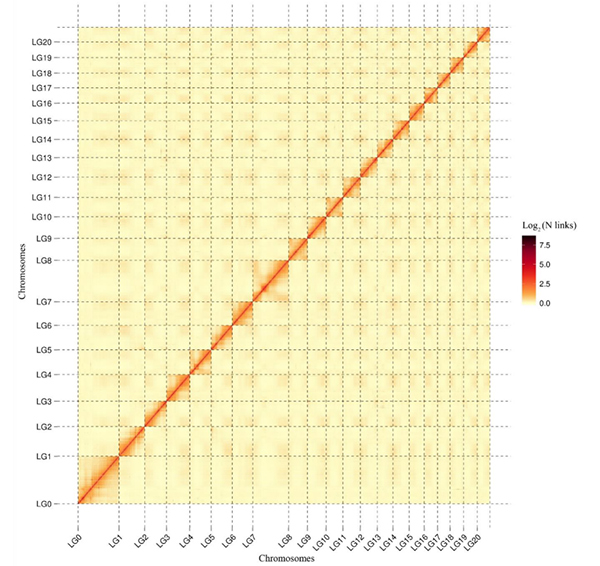
kang M et ai.,Luontoviestintä,2021
BMK kotelo
Laadukas genomikokonaisuus korostaa rukiin genomiominaisuuksia ja agronomisesti tärkeitä geenejä
Julkaistu: Luonnon genetiikka, 2021
Sekvensointistrategia:
Genomikokoonpano: PacBio CLR -tila 20 kb:n kirjastolla (497 Gt, noin 63×)
Sekvenssikorjaus: NGS 270 bp:n DNA-kirjastolla (430 Gb, noin 54×) Illumina-alustalla
Jatkoankkurointi: Hi-C-kirjasto (560 Gb, noin 71×) Illumina-alustalla
Optinen kartta: (779,55 Gb, noin 99×) Bionano Irysissä
Tärkeimmät tulokset
1. Weiningin rukiin genomin kokoonpano julkaistiin, jonka genomin kokonaiskoko oli 7,74 Gb (98,74 % arvioidusta genomin koosta virtaussytometrisesti).Tämän kokoonpanon teline N50 saavutti 1,04 Gb.93,67 % jatkuvista ankkuroitui onnistuneesti 7 pseudokromosomiin.Tämä kokoonpano arvioitiin linkage mapilla, LAI:lla ja BUSCO:lla, mikä johti korkeisiin pisteisiin kaikissa arvioinneissa.
2. Tämän genomin pohjalta suoritettiin lisätutkimuksia vertailevasta genomiikasta, geneettisen kytkentäkartan ja transkriptomiikan tutkimuksista.Paljastettiin joukko ominaisuuksiin liittyviä genomiominaisuuksia, mukaan lukien genomin laajuiset geenien kaksoiskappaleet ja niiden vaikutus tärkkelyksen biosynteesigeeneihin;monimutkaisten prolamiinilokusten fyysinen järjestäytyminen, geenin ilmentymisen ominaisuudet, jotka ovat taustalla varhaisen suuntausominaisuuden ja oletetut kesytykseen liittyvät kromosomialueet ja lokukset rukiissa.
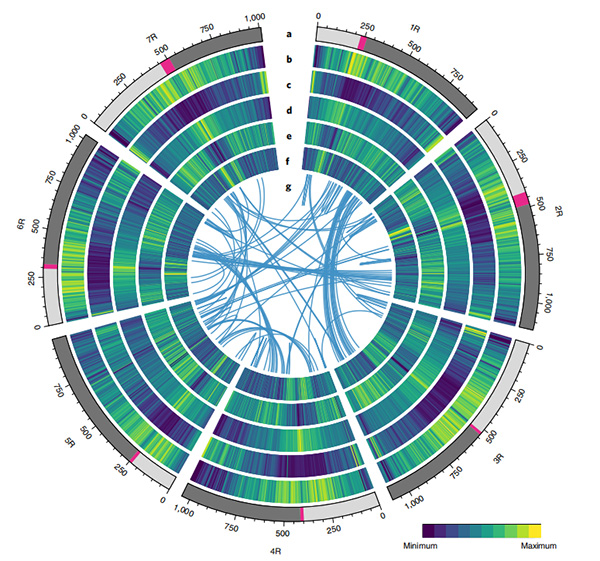 Circos-kaavio Weiningin rukiin genomin genomiominaisuuksista | 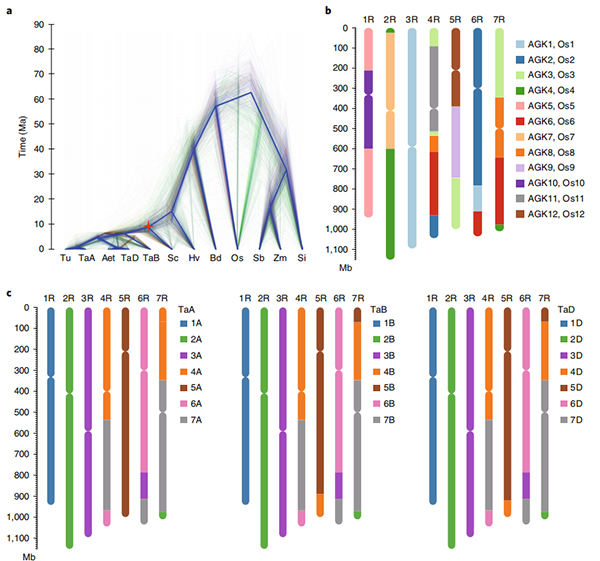 Rukiin genomin evoluutio- ja kromosomisynteenia-analyysit |
Li, G., Wang, L., Yang, J.et ai.Laadukas genomikokonaisuus korostaa rukiin genomiominaisuuksia ja agronomisesti tärkeitä geenejä.Nat Genet 53,574–584 (2021).
https://doi.org/10.1038/s41588-021-00808-z





