

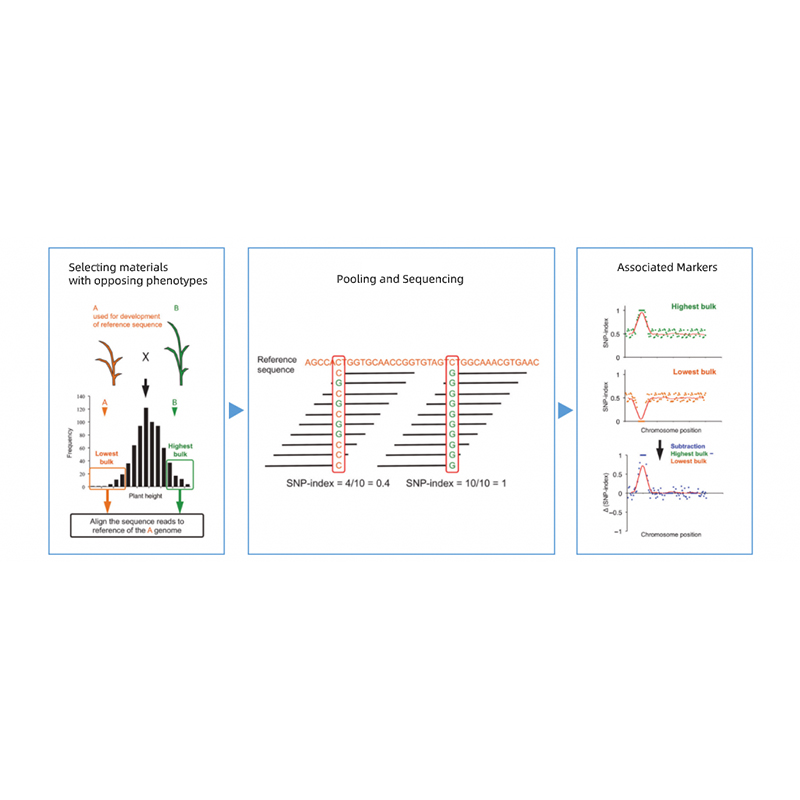
Analyse du ségrégant groupé






Avantages des services
Takagi et al., Le journal des plantes, 2013
● Localisation précise : Mélange de lots de 30+30 à 200+200 individus pour minimiser le bruit de fond ;prédiction de régions candidates non synonymes basée sur des mutants.
● Analyse complète : annotation approfondie de la fonction des gènes candidats, notamment NR, SwissProt, GO, KEGG, COG, KOG, etc.
● Délai d'exécution plus rapide : localisation rapide des gènes dans un délai de 45 jours ouvrables.
● Vaste expérience : BMK a contribué à la localisation de milliers de caractères, couvrant diverses espèces telles que les cultures, les produits aquatiques, les forêts, les fleurs, les fruits, etc.
Spécifications des services
Population:
Ségrégation de la descendance de parents présentant des phénotypes opposés.
Par exemple, descendance F2, rétrocroisement (BC), lignée consanguine recombinante (RIL)
Bassin de mélange
Pour les caractères qualitatifs : 30 à 50 individus (minimum 20)/vrac
Pour les tratis quantitatifs : les 5 à 10 % d'individus présentant l'un ou l'autre phénotype extrême dans l'ensemble de la population (minimum 30+30).
Profondeur de séquençage recommandée
Au moins 20X/parent et 1X/individu progéniture (par exemple, pour un pool de mélange de progéniture de 30+30 individus, la profondeur de séquençage sera de 30X par vrac)
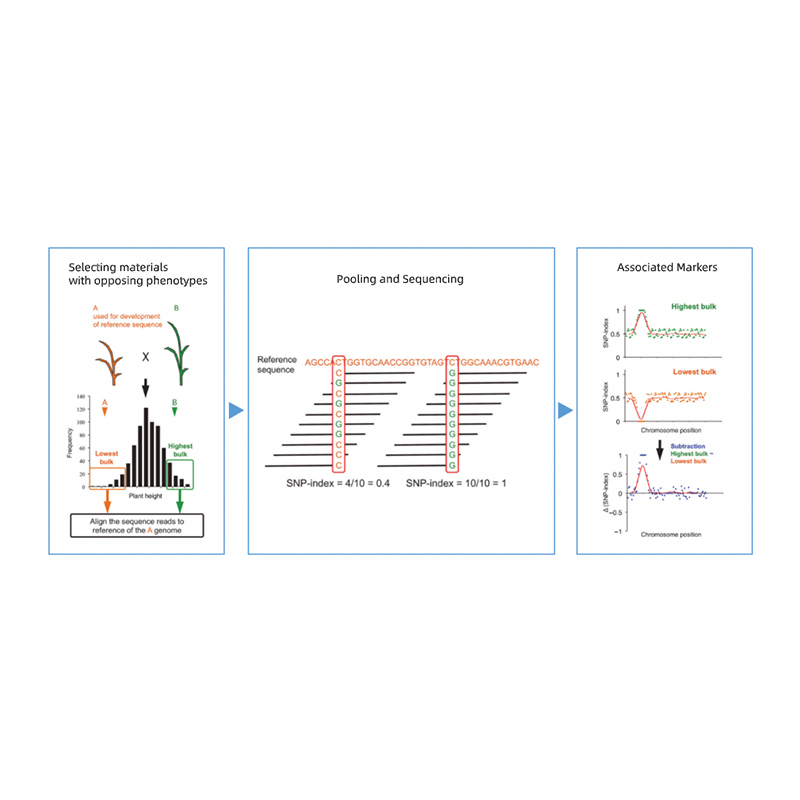
Analyses bioinformatiques
● Reséquençage du génome entier
● Traitement des données
● Appel SNP/Indel
● Sélection des régions candidates
● Annotation de la fonction du gène candidat
Exigences et livraison des échantillons
Exigences de l'échantillon :
Nucléotides :
| échantillon d'ADNg | Échantillon de tissu |
| Concentration : ≥30 ng/μl | Plantes : 1-2 g |
| Quantité : ≥2 μg (Volume ≥15 μl) | Animaux : 0,5-1 g |
| Pureté : OD260/280 = 1,6-2,5 | Sang total : 1,5 ml |
Flux de travail des services

Conception d'expériences

Livraison d'échantillon

Extraction d'ARN

Construction d'une bibliothèque

Séquençage

L'analyse des données

Services après-vente
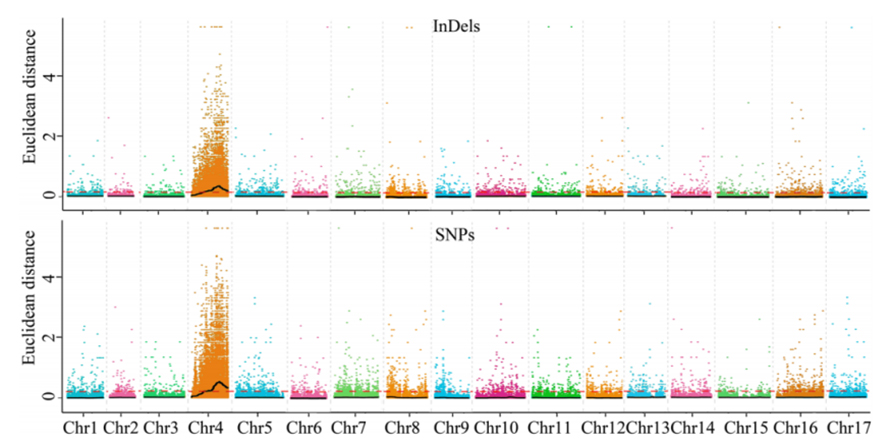
1.Base d'analyse d'association sur la distance euclidienne (ED) pour identifier la région candidate.Dans la figure suivante
Axe X : numéro de chromosome ;Chaque point représente une valeur ED d'un SNP.La ligne noire correspond à la valeur ED ajustée.Une valeur ED plus élevée indique une association plus significative entre le site et le phénotype.La ligne pointillée rouge représente le seuil d’association significative.
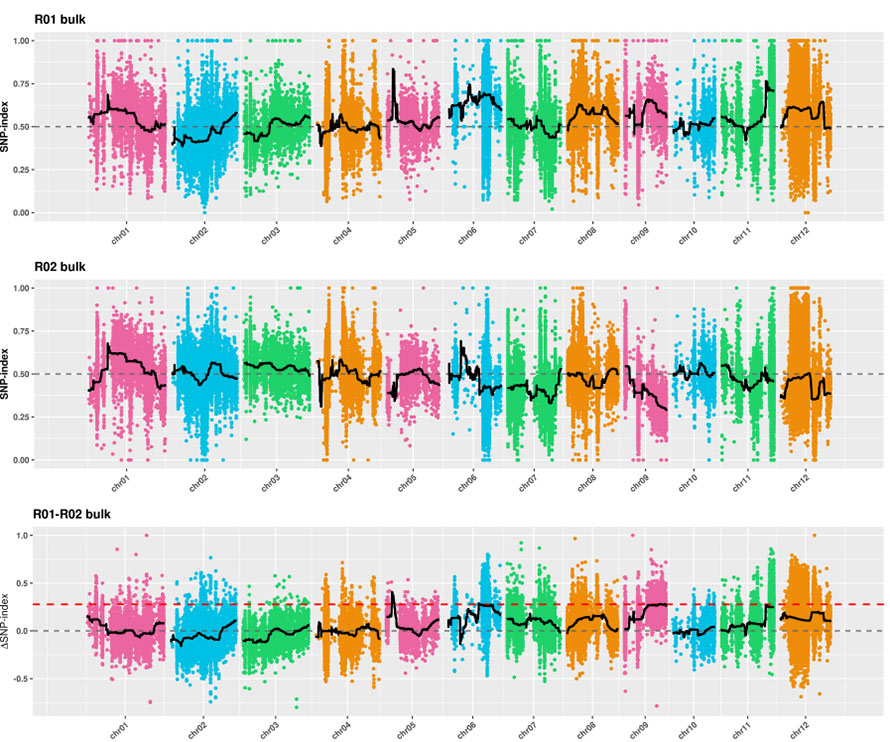
2. Analyse d'association basée sur aucun indice SNP
Axe X : numéro de chromosome ;Chaque point représente la valeur de l'indice SNP.La ligne noire représente la valeur de l'indice SNP ajustée.Plus la valeur est grande, plus l’association est significative.
Affaire BMK
Le locus du caractère quantitatif à effet majeur Fnl7.1 code pour une protéine abondante en fin d'embryogenèse associée à la longueur du cou du fruit chez le concombre.
Publié : Journal de biotechnologie végétale, 2020
Stratégie de séquençage :
Parents (Jin5-508, YN) : reséquençage du génome entier pour 34× et 20×.
Pools d'ADN (50 à col long et 50 à col court) : reséquençage pour 61× et 52×
Résultats clés
Dans cette étude, la population en ségrégation (F2 et F2: 3) a été générée en croisant la lignée de concombre à long cou Jin5-508 et la lignée de concombre à col court YN.Deux pools d'ADN ont été construits par 50 individus au cou extrêmement long et 50 individus au cou extrêmement court.Les QTL à effet majeur ont été identifiés sur Chr07 par analyse BSA et cartographie QTL traditionnelle.La région candidate a été encore réduite par une cartographie fine, une quantification de l'expression génique et des expériences transgéniques, qui ont révélé le gène clé dans le contrôle de la longueur du cou, CsFnl7.1.De plus, le polymorphisme dans la région promotrice CsFnl7.1 s’est avéré associé à l’expression correspondante.Une analyse phylogénétique plus approfondie suggère que le locus Fnl7.1 est très probablement originaire d'Inde.
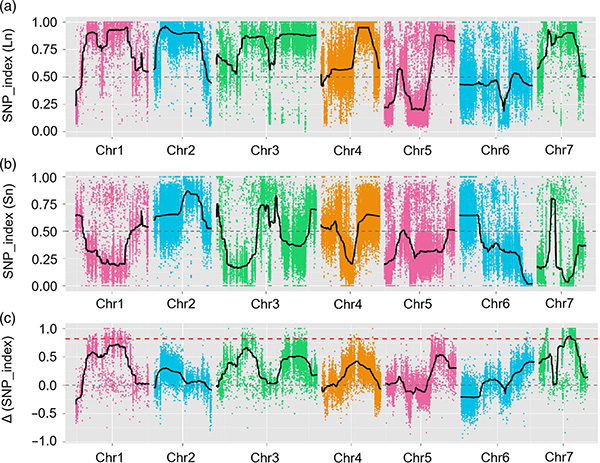 Cartographie QTL dans l'analyse BSA pour identifier la région candidate associée à la longueur du cou du concombre | 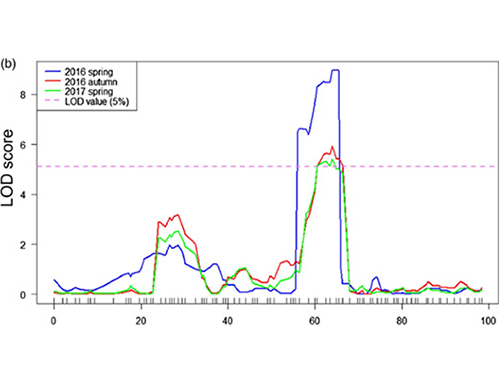 Profils LOD des QTL de longueur de cou de concombre identifiés sur Chr07 |
Xu, X. et coll."Le locus de caractère quantitatif à effet majeur Fnl7.1 code pour une protéine abondante en fin d'embryogenèse associée à la longueur du cou du fruit chez le concombre."Journal de biotechnologie végétale 18.7 (2020).





