

Hi-C alapú Genome Assembly







Szolgáltatás előnyei
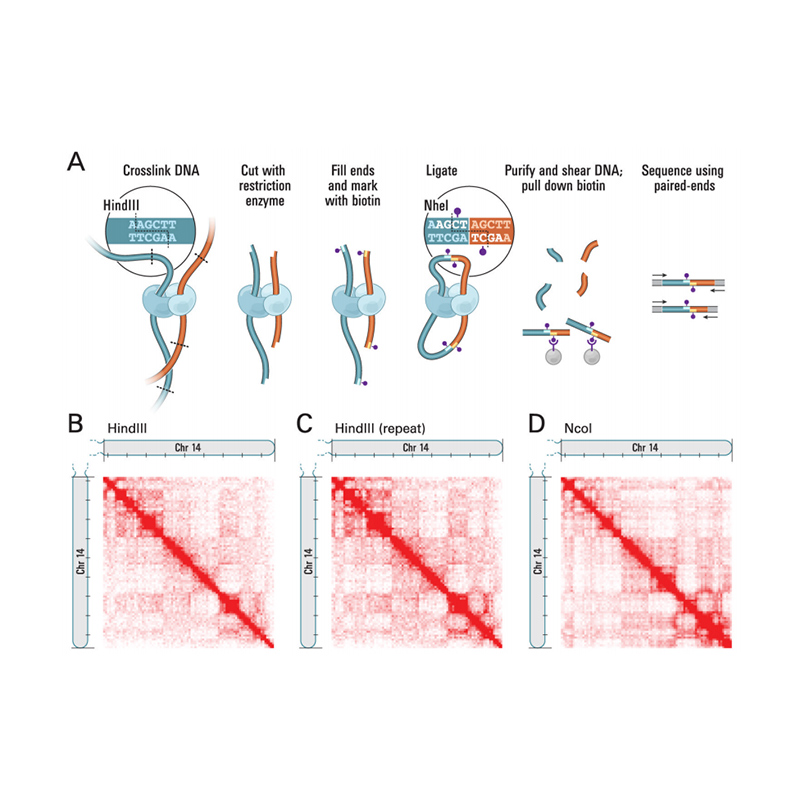
A Hi-C áttekintése
(Lieberman-Aiden E et al.,Tudomány, 2009)
● Nincs szükség genetikai populáció kialakítására a kontig horgonyzáshoz;
● Magasabb markersűrűség, ami magasabb kontig-rögzítési arányt eredményez 90% felett;
● Lehetővé teszi a meglévő genomegységek értékelését és korrekcióit;
● Rövidebb átfutási idő nagyobb pontossággal a genom összeállításban;
● Bőséges tapasztalat több mint 1000 Hi-C könyvtárral, amelyek több mint 500 faj számára készültek;
● Több mint 100 sikeres eset 760 feletti összesített publikált impakt faktorral;
● Hi-C alapú genom összeállítás poliploid genomhoz, az előző projektben 100%-os lehorgonyzási arányt sikerült elérni;
● Házon belüli szabadalmak és szoftver szerzői jogok Hi-C kísérletekhez és adatelemzésekhez;
● Saját fejlesztésű vizualizált adathangoló szoftver, amely lehetővé teszi a blokk kézi mozgatását, megfordítását, visszavonását és újrakészítését.
Szolgáltatási specifikációk
|
Könyvtár típusa
|
Felület | Olvassa el a hosszat | Stratégia ajánlása |
| Sziasztok | Illumina NovaSeq | PE150 | ≥ 100X |
Bioinformatikai elemzések
● Nyers adatok minőségének ellenőrzése
● Hi-C könyvtár minőségellenőrzése
● Hi-C alapú genom összeállítás
● Összeszerelés utáni értékelés
Mintakövetelmények és szállítás
Mintakövetelmények:
| Állat | Gomba | Növények
|
| Fagyasztott szövet: 1-2g könyvtáronként Sejtek: 1x 10^7 sejt könyvtáronként | Fagyasztott szövet: 1g könyvtáronként | Fagyasztott szövet: 1-2g könyvtáronként
|
| * Erősen javasoljuk, hogy küldjön legalább 2 alikvot részt (egyenként 1 g) a Hi-C kísérlethez. | ||
Javasolt mintaszállítás
Tartály: 2 ml-es centrifugacső (ón fólia nem ajánlott)
A legtöbb minta esetében azt javasoljuk, hogy ne tárolja etanolban.
Mintacímkézés: A mintákat egyértelműen fel kell címkézni, és meg kell egyeznie a benyújtott mintainformációs űrlappal.
Szállítás: Szárazjég: A mintákat először zsákokba kell csomagolni, és szárazjégbe kell temetni.
Szerviz munkafolyamat

Kísérleti tervezés

Mintaszállítás

DNS kivonás

Könyvtárépítés

Szekvenálás

Adatelemzés

Értékesítés utáni szolgáltatások
*Az itt látható demo eredmények mind a Biomarker Technologies által közzétett genomokból származnak
1.Hi-C kölcsönhatás hőtérképeCamptotheca acuminatagenom.Amint a térképen látható, az interakciók intenzitása negatívan korrelál a lineáris távolsággal, ami nagyon pontos kromoszóma szintű összeállítást jelez.(Rögzítési arány: 96,03%)
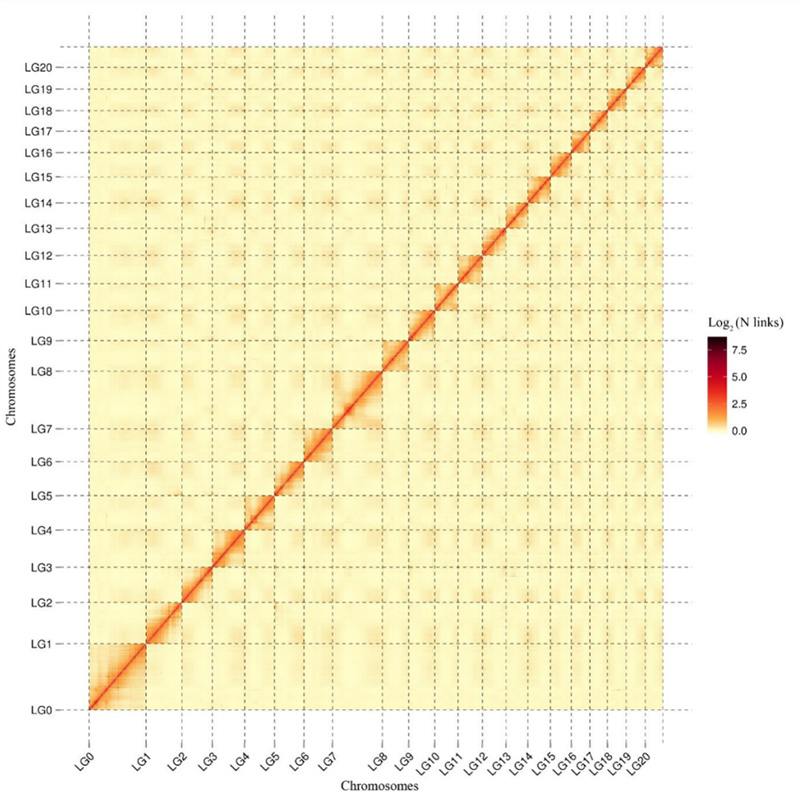
Kang M et al.,Nature Communications, 2021
2. A Hi-C megkönnyítette a közötti inverziók érvényesítésétGossypium hirsutumL. TM-1 A06 ésG. arboreumChr06
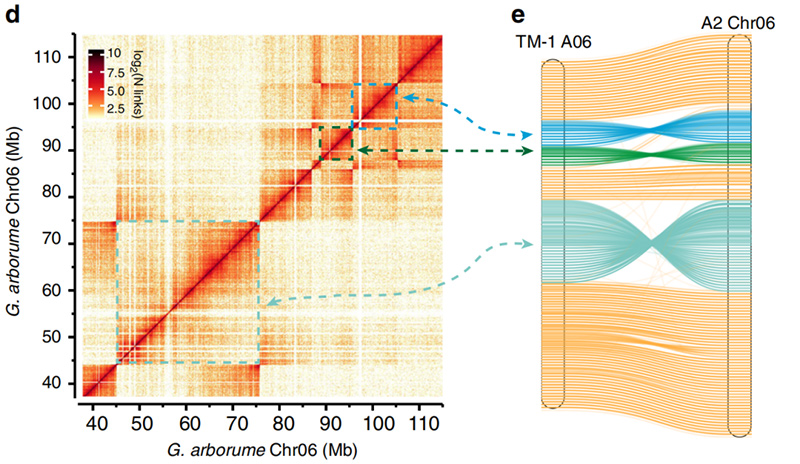
Yang Z et al.,Nature Communications, 2019
3. Az SC205 manióka genom összeállítása és biallélikus differenciálódása.A Hi-C hőtérkép egyértelmű hasadást mutat a homológ kromoszómákban.
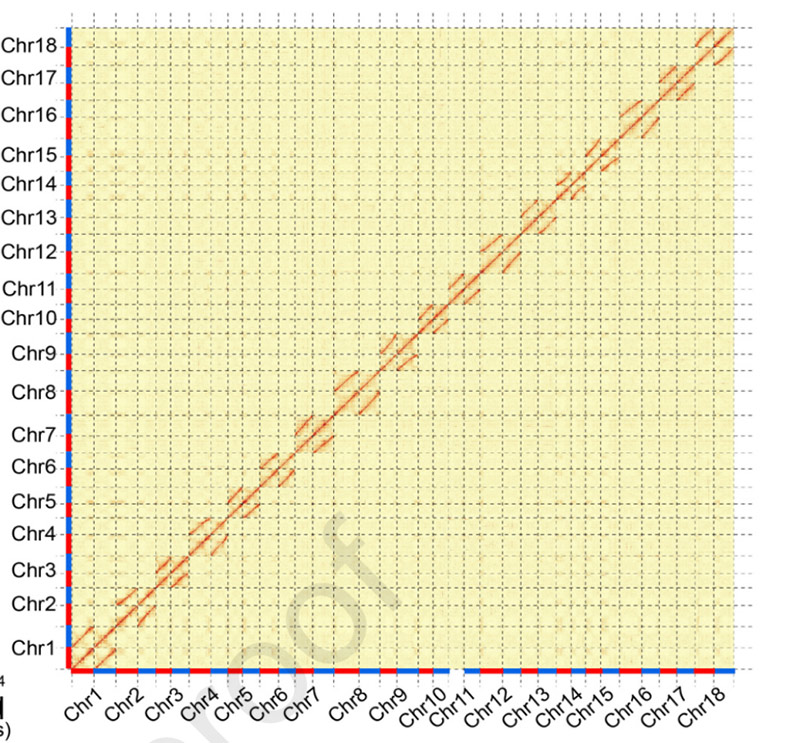
Hu W et al.,Molekuláris növény, 2021
4. Hi-C hőtérkép két Ficus faj genom összeállításán:F.microcarpa(lehorgonyzási arány: 99,3%) illF.hispida (rögzítési arány: 99,7%)
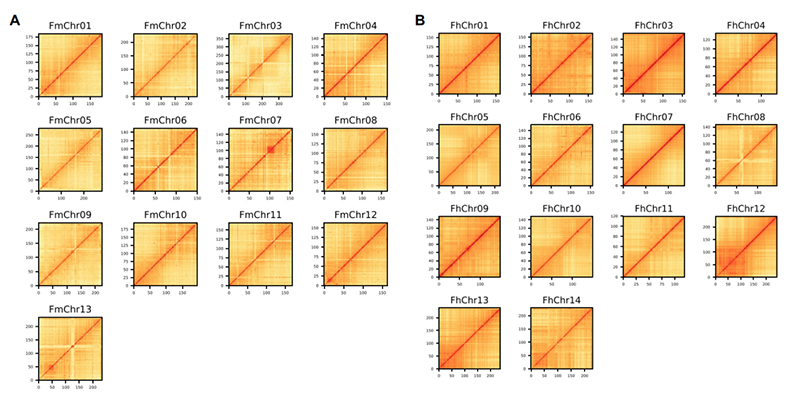
Zhang X és társai,Sejt, 2020
BMK tok
A banyánfa és a beporzó darázs genomja betekintést nyújt a füge-darázs koevolúciójába
Közzétett: Sejt, 2020
Szekvenálási stratégia:
F. microcarpa genom: kb.84 X PacBio RSII (36,87 Gb) + Hi-C (44 Gb)
F. hispidagenom: kb.97 X PacBio RSII (36,12 Gb) + Hi-C (60 Gb)
Eupristina verticillatagenom: kb.170 X PacBio RSII (65 Gb)
Főbb eredmények
1. Két banyan fa genomot és egy beporzó darázs genomot állítottunk elő PacBio szekvenálás, Hi-C és kapcsolódási térkép segítségével.
(1)F. microcarpagenom: 426 Mb (a becsült genomméret 97,7%-a) összeállítást hoztunk létre 908 Kb N50 kontiggel, 95,6%-os BUSCO pontszámmal.A Hi-C összesen 423 Mb szekvenciát rögzített 13 kromoszómához.A genom annotáció 29 416 fehérjét kódoló gént eredményezett.
(2)F. Hispidagenom: 360 Mb-os összeállítás (a becsült genomméret 97,3%-a) 492 Kb kontig N50 és 97,4% BUSCO pontszám mellett adódott.A Hi-C összesen 359 Mb méretű szekvenciát rögzített 14 kromoszómán, és nagymértékben azonos a nagy sűrűségű kapcsolódási térképpel.
(3)Eupristina verticillatagenom: Egy 387 Mb-os összeállítást (becsült genomméret: 382 Mb) hoztunk létre 3,1 Mb N50-vel és 97,7%-os BUSCO-pontszámmal.
2. Az összehasonlító genomikai elemzés nagyszámú szerkezeti eltérést tárt fel a kettő közöttFicusgenomokat, amelyek felbecsülhetetlen értékű genetikai erőforrást biztosítottak az adaptív evolúciós vizsgálatokhoz.Ez a tanulmány először nyújtott betekintést a fügedarázs genomi szintű koevolúciójába.
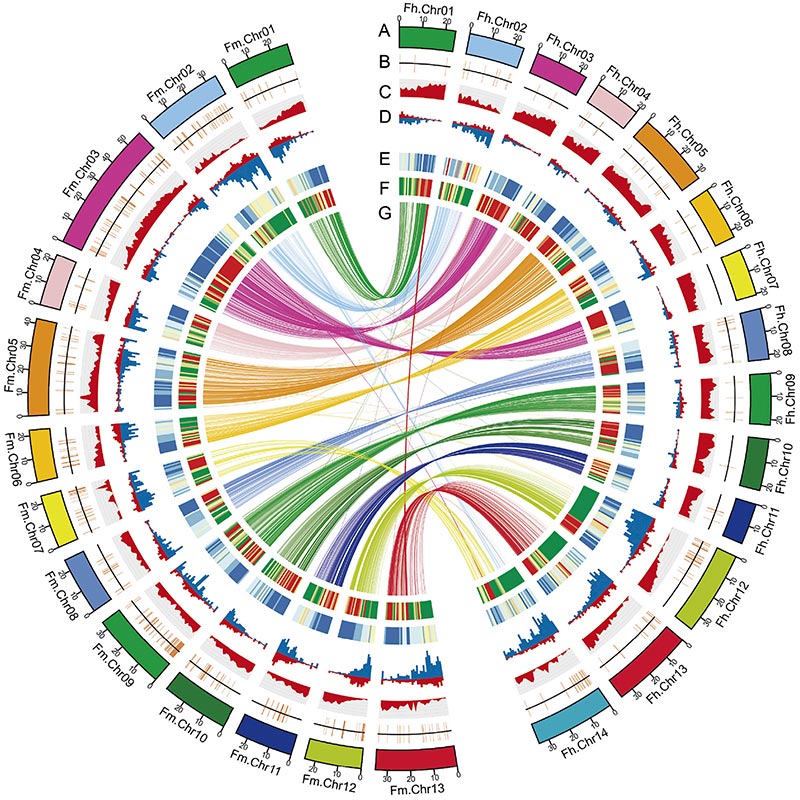 Circos diagram két genomikai jellemzőirőlFicusgenomok, beleértve a kromoszómákat, szegmentális duplikációkat (SD-ket), transzpozonokat (LTR, TE-k, DNS TE-k), génexpressziót és szinténiát | 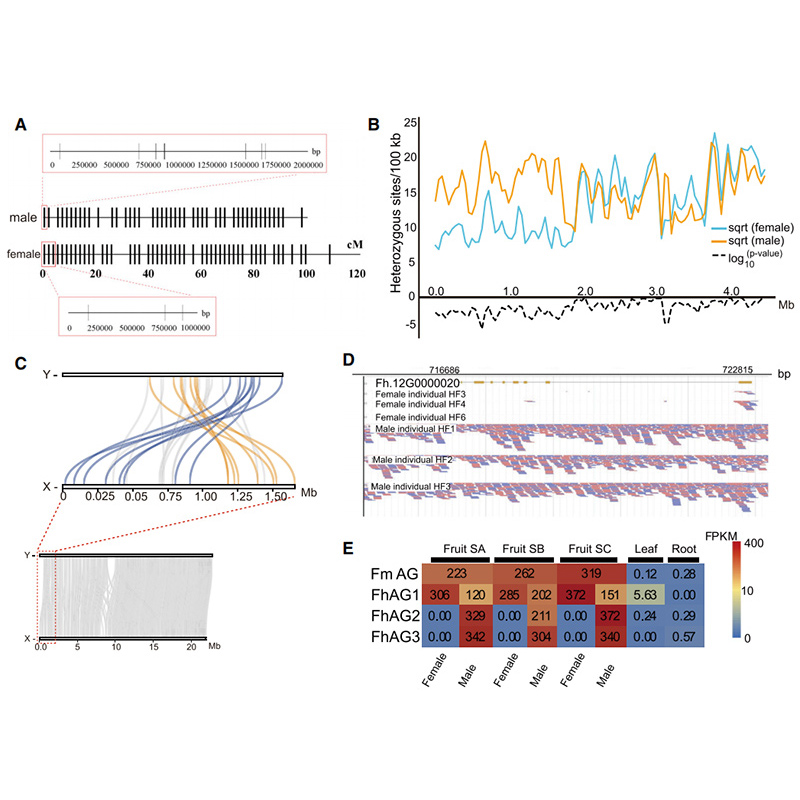 Az Y kromoszóma és az ivarmeghatározó jelölt gén azonosítása |
Zhang, X. és mtsai.„A banyánfa és a beporzó darázs genomjai betekintést nyújtanak a füge-darázs koevolúciójába.”183.4 (2020) cella.





