Emberi egész exome szekvenálás




Szolgáltatás előnyei
● Célzott fehérjekódoló régió: a fehérjét kódoló régió rögzítésével és szekvenálásával a hWES a fehérjeszerkezettel kapcsolatos variánsok feltárására szolgál;
● Nagy pontosság: a nagy szekvenálási mélységgel a hWES megkönnyíti az 1%-nál alacsonyabb frekvenciájú gyakori és ritka változatok észlelését;
● Költséghatékony: a hWES az emberi betegség mutációinak körülbelül 85%-át hozza létre az emberi genom 1%-ából;
● Öt szigorú minőségellenőrzési eljárás, amelyek lefedik a teljes folyamatot, Q30>85% garantált.
Minta specifikációk
| Felület
| Könyvtár
| Exon rögzítési stratégia
| Szekvenálási stratégia ajánlása
|
|
Illumina NovaSeq platform
| PE150 | Agilent SureSelect Human All Exon V6 IDT xGen Exome Hyb Panel V2 | 5 Gb 10 Gb |
Mintakövetelmények
| Minta típusa
| Összeg(Qubit®)
| Hangerő
| Koncentráció
| Tisztaság (NanoDrop™) |
|
Genomi DNS
| ≥ 300 ng | ≥ 15 μL | ≥ 20 ng/μL | OD260/280=1,8-2,0 nincs lebomlás, nincs szennyeződés
|
Ajánlott szekvenálási mélység
Mendeli rendellenességek/ritka betegségek esetén: effektív szekvenálási mélység 50× felett
Tumorminták esetén: effektív szekvenálási mélység 100× felett
Bioinformatika
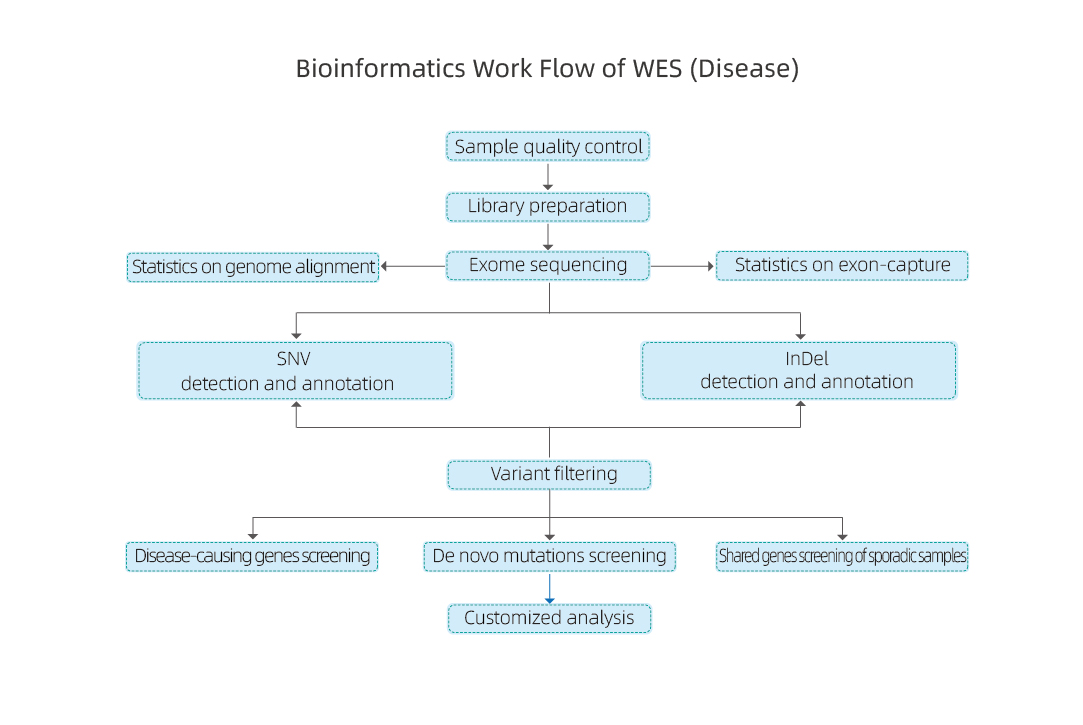
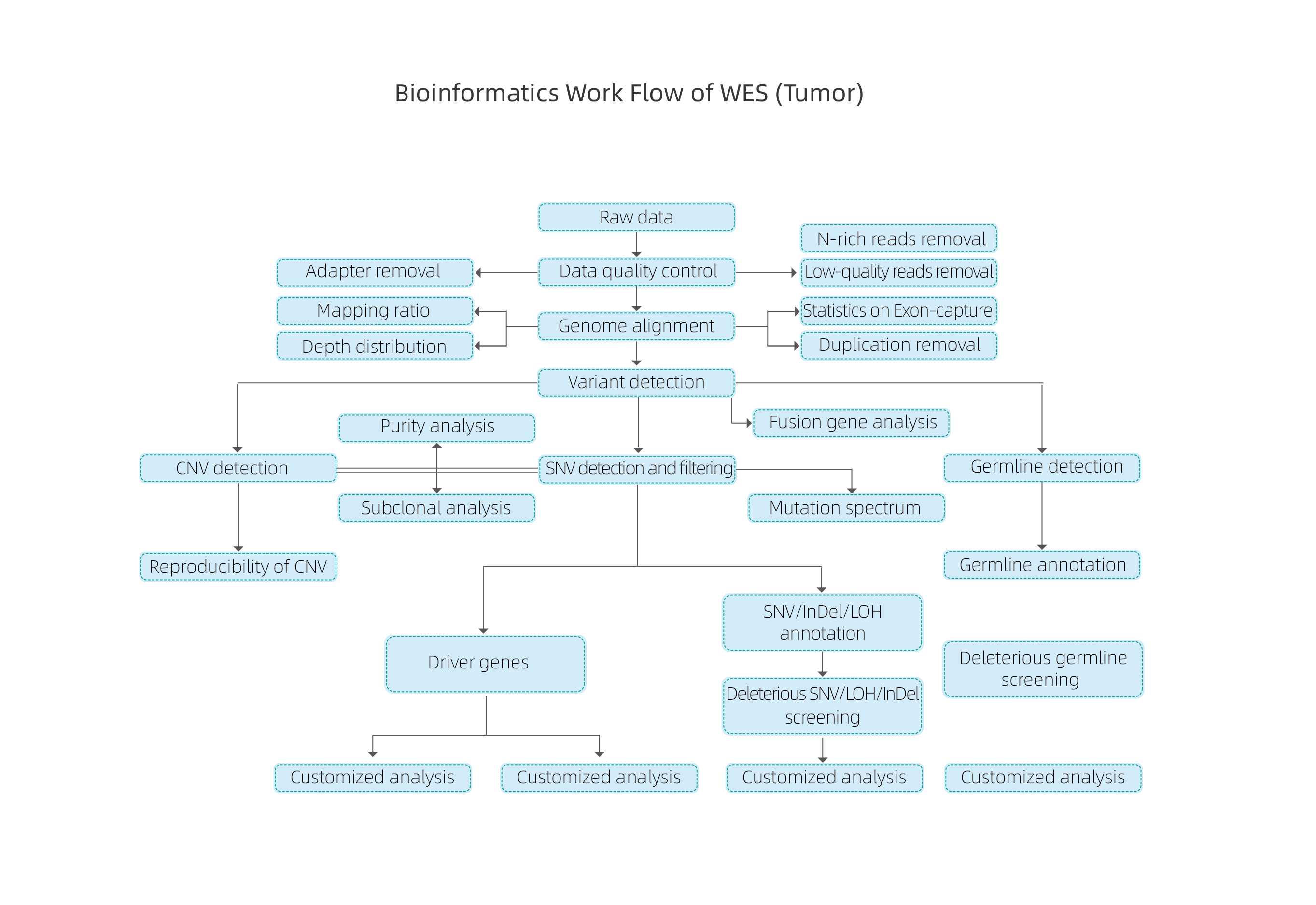
Szerviz munkafolyamat
Mintaszállítás

DNS kivonás

Könyvtárépítés

Szekvenálás

Adatelemzés
Adatszolgáltatás

Értékesítés utáni szolgáltatások
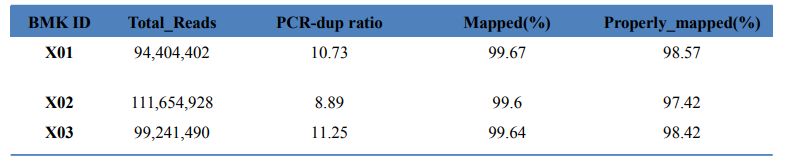
1. Igazítási statisztikák
1. táblázat A térképeredmény statisztikája
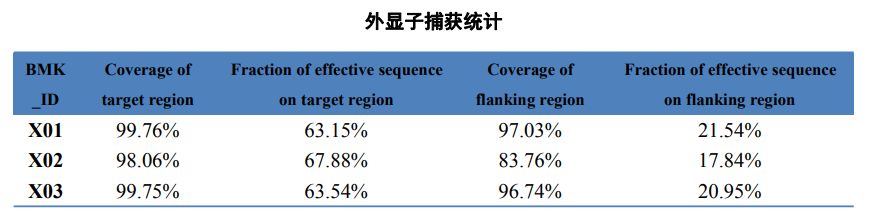
2. táblázat Az exome capture statisztikái
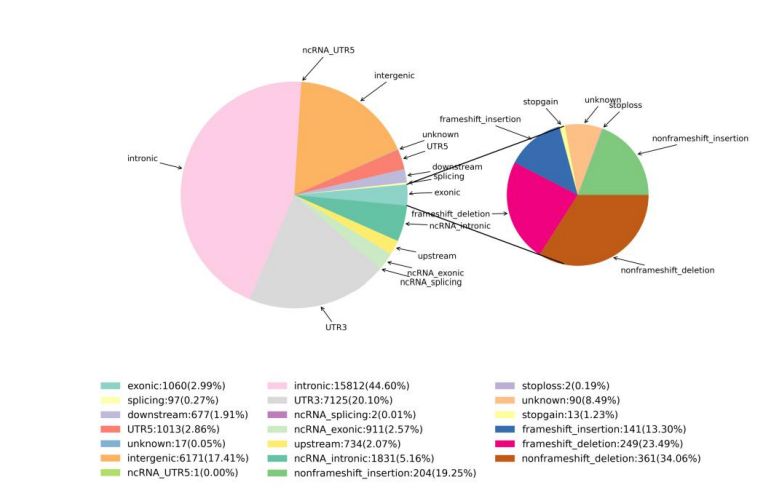
2. Változások észlelése
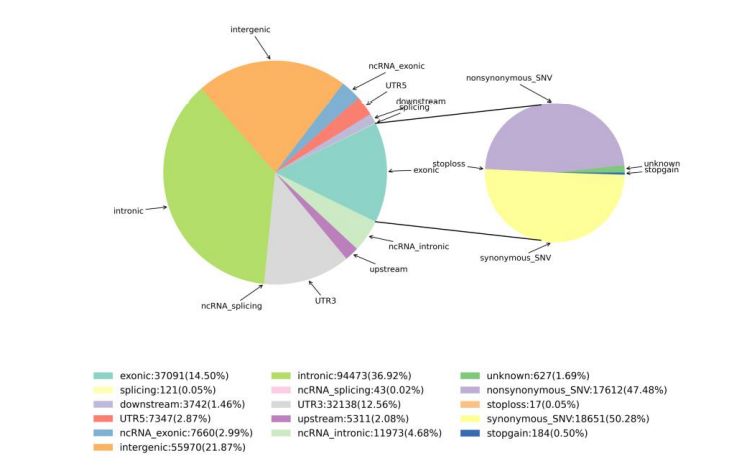
1. ábra Az SNV és az InDel statisztikái
3. Speciális elemzés
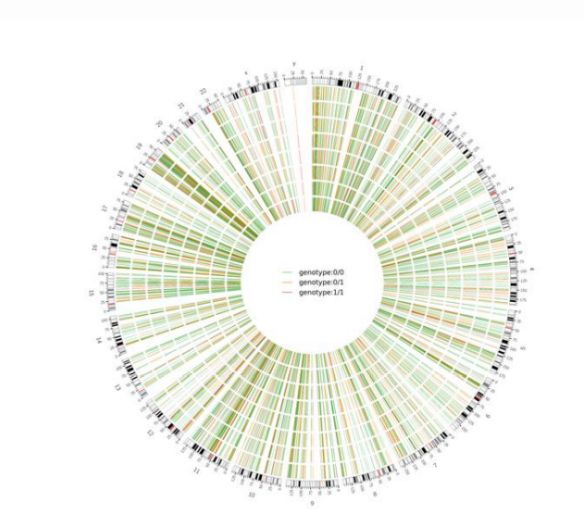
2. ábra Circos diagram a genom-szerte káros SNV és InDel
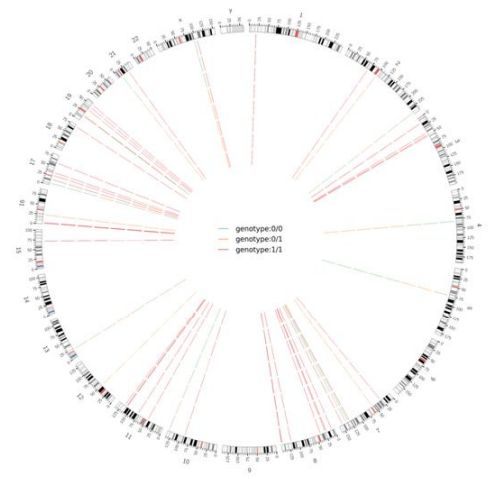
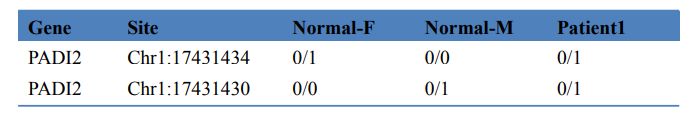
3. táblázat Betegséget okozó gének szűrése