

Hi-Cベースのゲノムアセンブリ







サービスのメリット
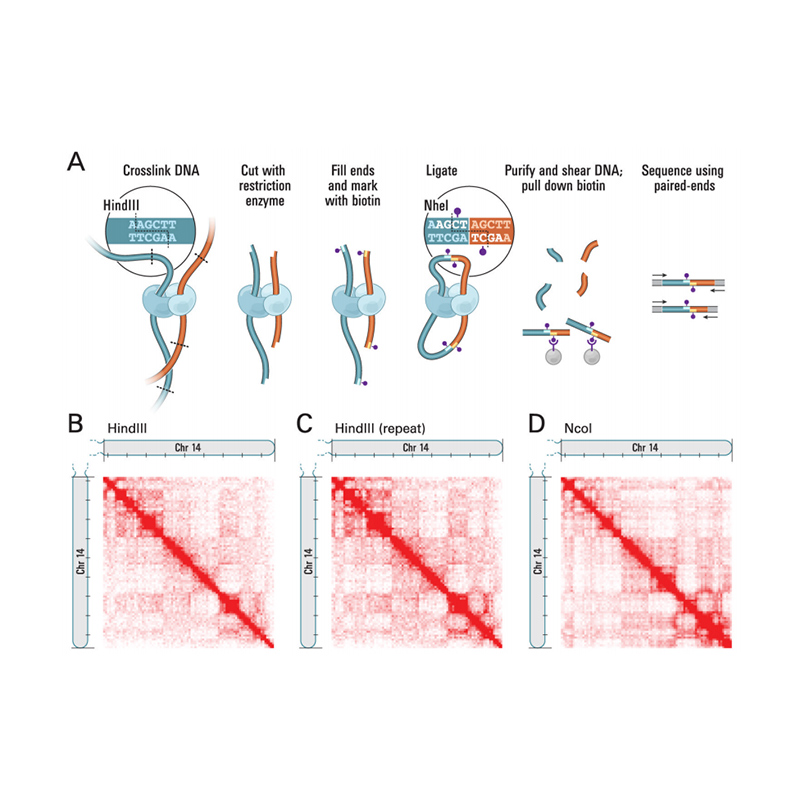
Hi-Cの概要
(リーバーマン-エイデン E ら、科学、2009)
● コンティグアンカーリングのための遺伝子集団を構築する必要はありません。
● マーカー密度が高いため、90% 以上のコンティグ固定率が高くなります。
● 既存のゲノムアセンブリの評価と修正を可能にします。
● ゲノムアセンブリの精度が高く、所要時間が短縮されます。
● 500 種を超える種に対して構築された 1000 を超える Hi-C ライブラリに関する豊富な経験。
● 累計公開インパクトファクターが 760 以上の 100 件を超える成功事例。
● Hi-C ベースの倍数体ゲノムのゲノムアセンブリ。以前のプロジェクトで 100% のアンカー率が達成されました。
● Hi-C 実験およびデータ分析に関する社内特許およびソフトウェア著作権。
● 自社開発の可視化データチューニングソフトウェアにより、手動でブロックの移動、反転、取り消し、やり直しが可能です。
サービス仕様
|
ライブラリの種類
|
プラットホーム | 読み取り長 | 推奨戦略 |
| ハイシー | イルミナ ノヴァシーク | PE150 | ≥ 100X |
バイオインフォマティクス分析
●生データの品質管理
● Hi-Cライブラリの品質管理
● Hi-Cベースのゲノムアセンブリ
●組み立て後の評価
サンプルの要件と納品
サンプル要件:
| 動物 | 真菌 | 植物
|
| 凍結組織: ライブラリあたり 1 ~ 2g セル: ライブラリあたり 1x 10^7 セル | 凍結組織: ライブラリあたり 1g | 凍結組織: ライブラリあたり 1 ~ 2g
|
| *Hi-C 実験には少なくとも 2 つのアリコート (各 1 g) を送ることを強くお勧めします。 | ||
推奨されるサンプル配信
容器: 2 ml 遠沈管 (錫箔は推奨しません)
ほとんどのサンプルは、エタノール中で保存しないことをお勧めします。
サンプルのラベル付け: サンプルには明確にラベルが付けられ、提出されたサンプル情報フォームと同一である必要があります。
発送: ドライアイス: サンプルは最初にバッグに梱包し、ドライアイスに埋める必要があります。
サービスのワークフロー

実験計画

サンプル納品

DNA抽出

図書館の建設

シーケンス

データ分析

アフターサービス
*ここに示されているデモの結果はすべて、Biomarker Technologies で公開されているゲノムからのものです
1.Hi-C相互作用ヒートマップ尖形カンプトテカゲノム。地図に示されているように、相互作用の強度は直線距離と負の相関があり、染色体レベルでのアセンブリが高精度に行われていることを示しています。(定着率:96.03%)
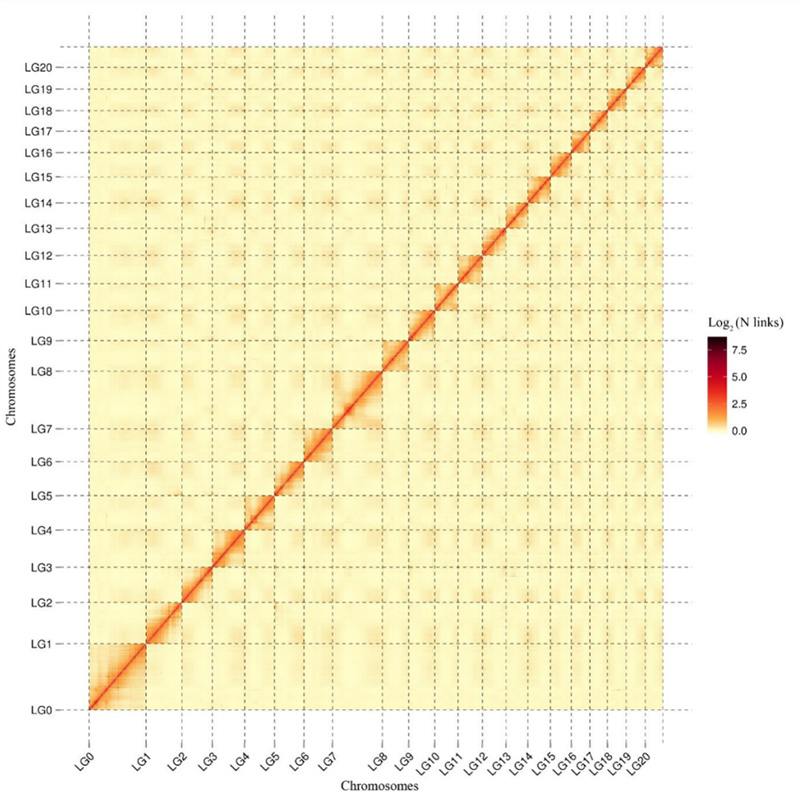
カン・Mら、ネイチャーコミュニケーションズ、2021年
2.Hi-C は、間の反転の検証を容易にしました。ゴシピウム・ヒルスタムL. TM-1 A06 およびG.樹木園Chr06
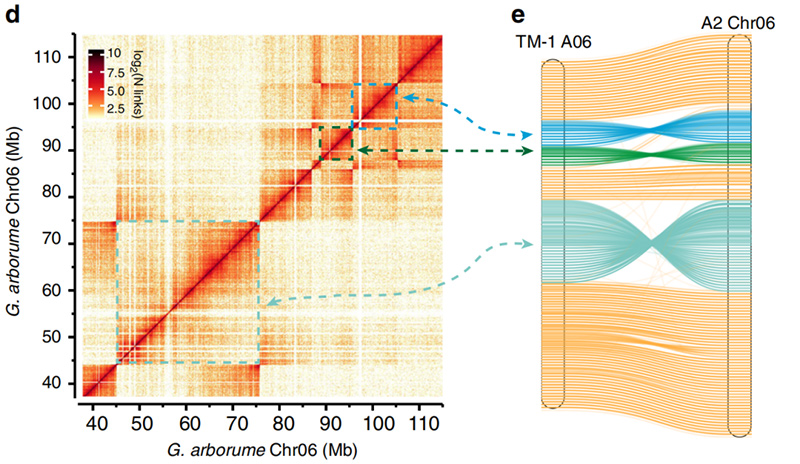
ヤン・Zら、ネイチャーコミュニケーションズ、2019
3.キャッサバゲノムSC205の構築と二対立遺伝子の分化。Hi-C ヒートマップでは、相同染色体の明確な分割が示されました。
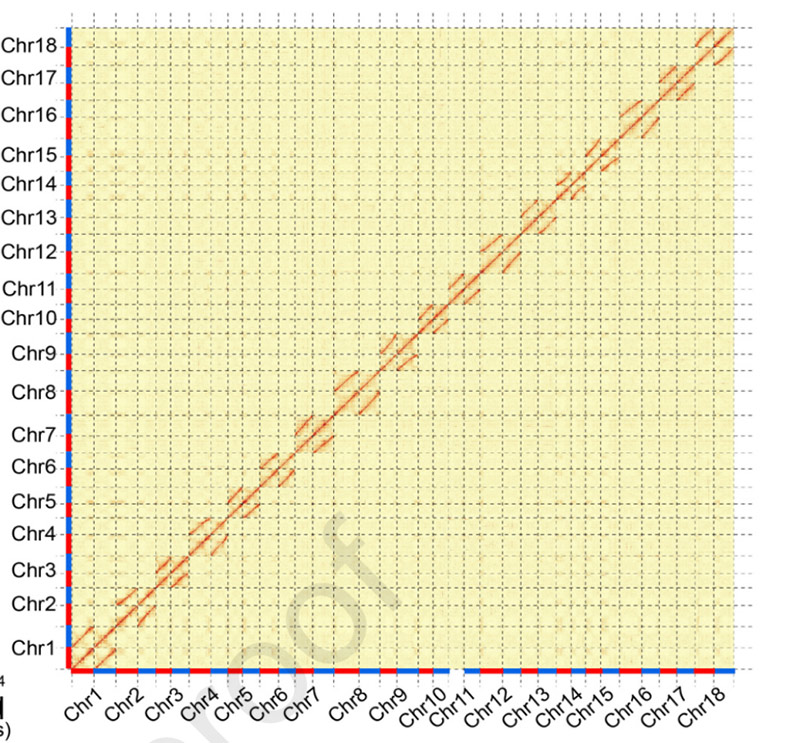
胡Wら、分子植物、2021年
4. 2 つのフィカス種ゲノム アセンブリの Hi-C ヒートマップ:F.ミクロカルパ(定着率99.3%)とF.hispida(定着率99.7%)
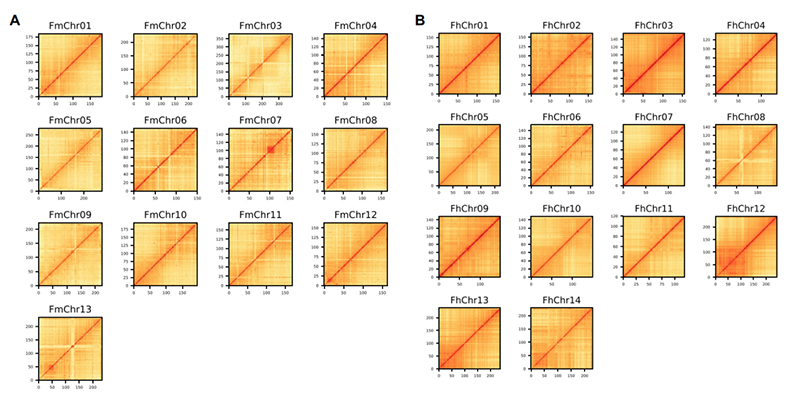
Zhang Xら、細胞、2020年
BMKケース
ガジュマルの木と花粉媒介バチのゲノムからイチジクとスズメバチの共進化に関する洞察が得られる
公開日: 細胞、2020年
シーケンス戦略:
F. マイクロカルパ ゲノム: 約。84 X PacBio RSII (36.87 Gb) + Hi-C (44 Gb)
F.ヒスピダゲノム: 約。97 X PacBio RSII (36.12 Gb) + Hi-C (60 Gb)
エウプリシュティナ バーティシラータゲノム: 約。170 X PacBio RSII (65 GB)
主な結果
1.2つのガジュマルゲノムと1つの花粉媒介ハチゲノムを、PacBioシーケンス、Hi-Cおよび連鎖マップを使用して構築しました。
(1)F. マイクロカルパゲノム: 426 Mb (推定ゲノム サイズの 97.7%) のアセンブリが、908 Kb のコンティグ N50、95.6% の BUSCO スコアで確立されました。合計 423 Mb 配列が Hi-C によって 13 染色体に固定されました。ゲノム注釈により、29,416 個のタンパク質をコードする遺伝子が得られました。
(2)F.ヒスピダゲノム: 360 Mb (推定ゲノム サイズの 97.3%) のアセンブリが収量され、コンティグ N50 は 492 Kb、BUSCO スコアは 97.4% でした。合計 359 Mb 配列が Hi-C によって 14 染色体上に固定されており、高密度連鎖マップと高度に同一でした。
(3)エウプリシュティナ バーティシラータゲノム: 387 Mb (推定ゲノム サイズ: 382 Mb) のアセンブリが、3.1 Mb のコンティグ N50 および 97.7% の BUSCO スコアで確立されました。
2.比較ゲノミクス分析により、2つの間の非常に多くの構造の違いが明らかになりました。イチジクゲノムは、適応進化の研究に貴重な遺伝資源を提供しました。この研究は、イチジクとスズメバチの共進化についてのゲノムレベルでの洞察を初めて提供した。
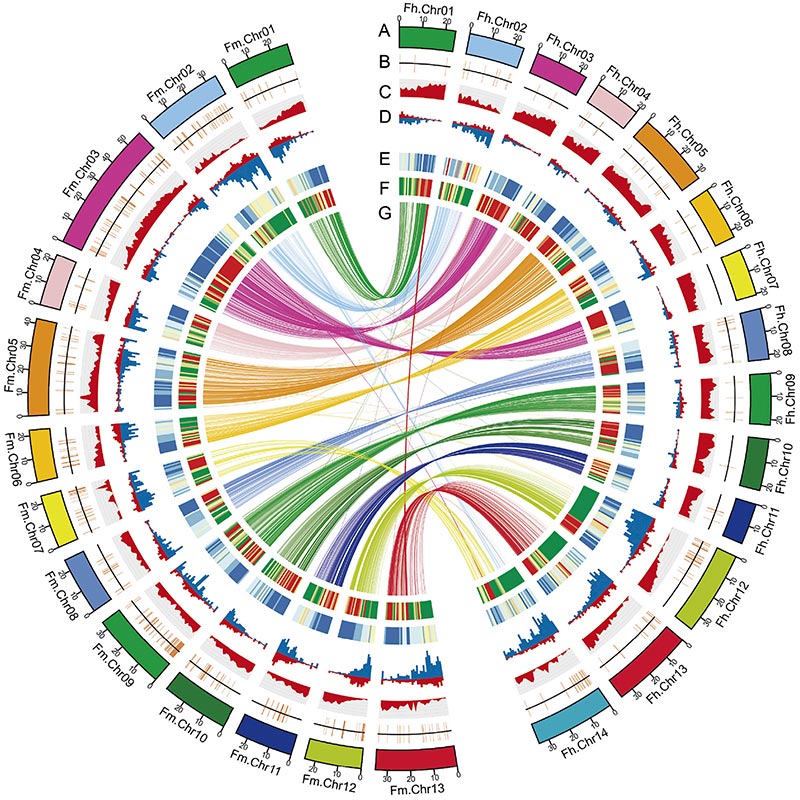 2人のゲノム特徴に関するCircos図イチジク染色体、分節重複 (SD)、トランスポゾン (LTR、TE、DNA TE)、遺伝子発現およびシンテニーを含むゲノム | 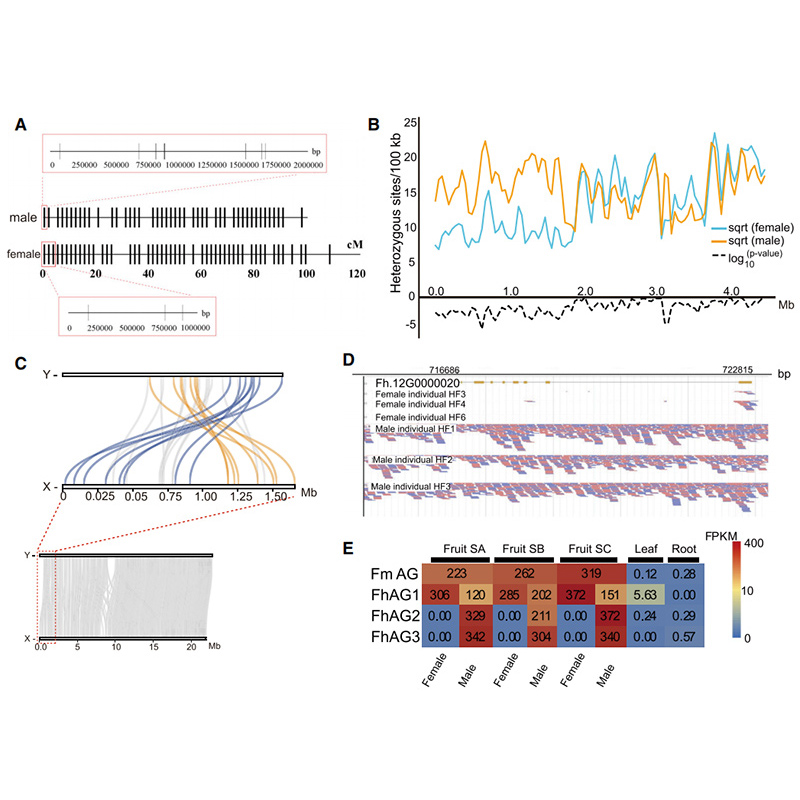 Y染色体と性決定候補遺伝子の同定 |
Zhang、X.ら。「ガジュマルの木と花粉媒介のスズメバチのゲノムは、イチジクとスズメバチの共進化に関する洞察を提供します。」セル183.4(2020)。





