-
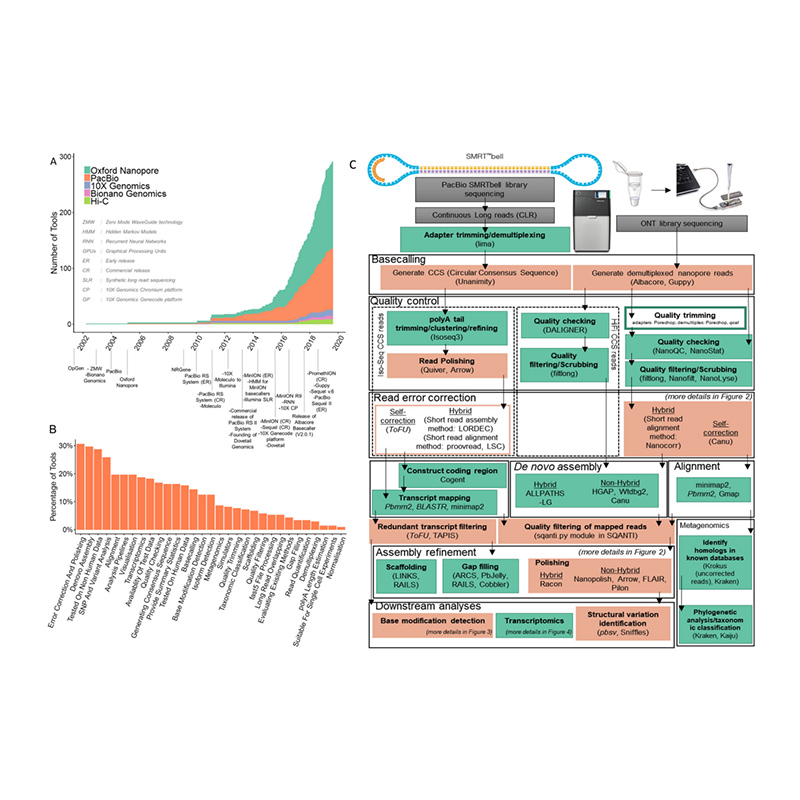
ການຈັດລຽງລຳດັບພັນທຸກໍາຂອງພືດ/ສັດ
ເດໂນໂວການຈັດລຳດັບໝາຍເຖິງການສ້າງ genome ທັງໝົດຂອງຊະນິດພັນໂດຍໃຊ້ເທັກໂນໂລຍີການຈັດລຳດັບ, ເຊັ່ນ PacBio, Nanopore, NGS, ແລະອື່ນໆ, ໃນເມື່ອບໍ່ມີ genome ອ້າງອີງ.ການປັບປຸງທີ່ໂດດເດັ່ນໃນຄວາມຍາວການອ່ານຂອງເຕັກໂນໂລຊີການຈັດລໍາດັບລຸ້ນທີສາມໄດ້ນໍາເອົາໂອກາດໃຫມ່ໃນການປະກອບ genomes ທີ່ຊັບຊ້ອນ, ເຊັ່ນວ່າຜູ້ທີ່ມີ heterozygosity ສູງ, ອັດຕາສ່ວນສູງຂອງພາກພື້ນທີ່ຊ້ໍາກັນ, polyploids, ແລະອື່ນໆ. ດ້ວຍຄວາມຍາວການອ່ານໃນລະດັບຫຼາຍສິບກິໂລຖານ, ການຈັດລໍາດັບເຫຼົ່ານີ້ສາມາດອ່ານໄດ້. ການແກ້ໄຂອົງປະກອບທີ່ຊ້ໍາ, ພາກພື້ນທີ່ມີເນື້ອໃນ GC ຜິດປົກກະຕິແລະພາກພື້ນທີ່ສະລັບສັບຊ້ອນສູງອື່ນໆ.
ເວທີ: PacBio Sequel II / Nanopore PromethION P48 / Illumina NovaSeq6000
-
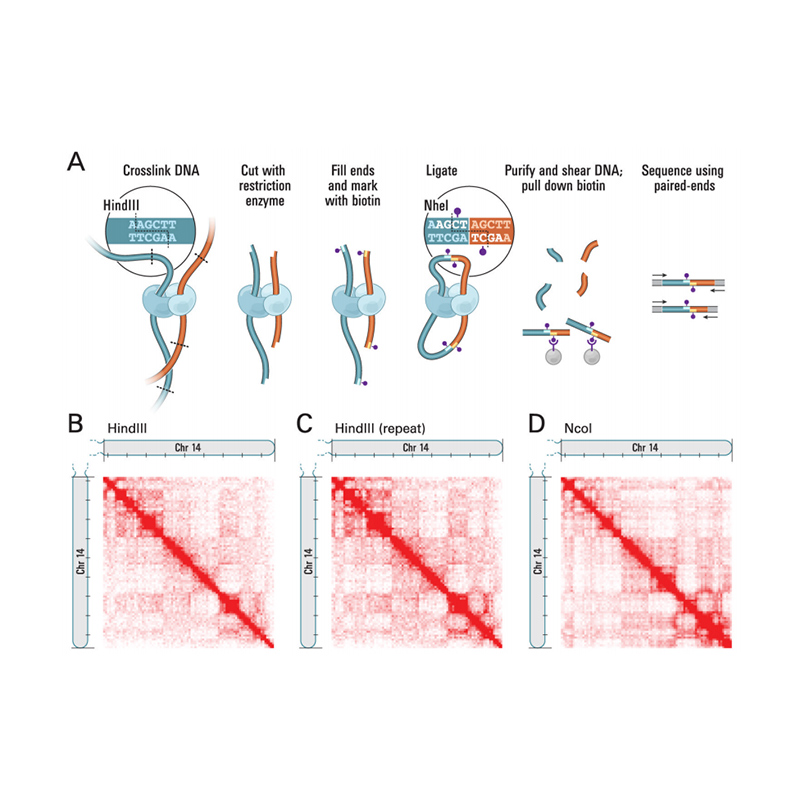
ສະພາແຫ່ງ Genome ທີ່ອີງໃສ່ Hi-C
Hi-C ແມ່ນວິທີການທີ່ອອກແບບມາເພື່ອບັນທຶກການຕັ້ງຄ່າໂຄໂມໂຊມໂດຍການລວມເອົາການໂຕ້ຕອບທີ່ອີງໃສ່ຄວາມໃກ້ຄຽງ ແລະການຈັດລໍາດັບການສົ່ງຜ່ານສູງ.ຄວາມເຂັ້ມຂຸ້ນຂອງປະຕິສໍາພັນເຫຼົ່ານີ້ຖືກເຊື່ອວ່າມີຄວາມສໍາພັນທາງລົບກັບໄລຍະຫ່າງທາງດ້ານຮ່າງກາຍຢູ່ໃນໂຄໂມໂຊມ.ດັ່ງນັ້ນ, ຂໍ້ມູນ Hi-C ສາມາດແນະນຳການຈັດກຸ່ມ, ການສັ່ງ ແລະ ການກຳນົດທິດທາງຂອງລຳດັບທີ່ປະກອບຢູ່ໃນ genome ສະບັບຮ່າງ ແລະ ຍຶດເອົາໂຄໂມໂຊມຈຳນວນທີ່ແນ່ນອນ.ເທກໂນໂລຍີນີ້ສ້າງຄວາມເຂັ້ມແຂງການປະກອບ genome ໃນລະດັບໂຄໂມໂຊມໃນກໍລະນີທີ່ບໍ່ມີແຜນທີ່ພັນທຸກໍາໂດຍອີງໃສ່ປະຊາກອນ.ທຸກໆ genome ຕ້ອງການ Hi-C.
ເວທີ: Illumina NovaSeq6000 / DNBSEQ
-
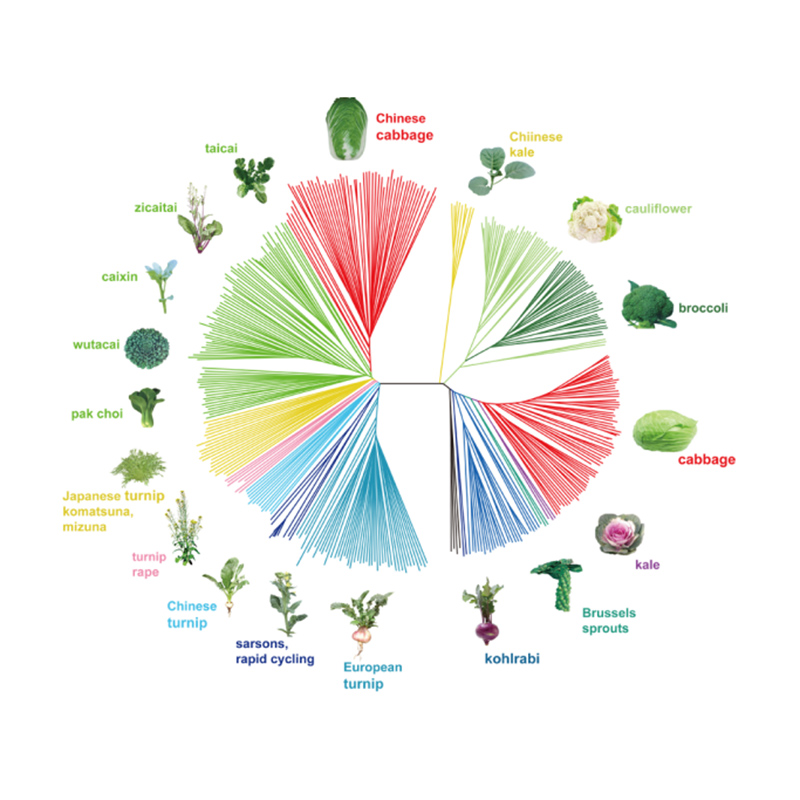
ພັນທຸ ກຳ ວິວັດທະນາການ
ພັນທຸ ກຳ Evolutionary ແມ່ນການບໍລິການຈັດລຽງຕາມລຳດັບທີ່ອອກແບບມາເພື່ອສະໜອງການຕີຄວາມສົມບູນກ່ຽວກັບຂໍ້ມູນວິວັດທະນາການຂອງວັດສະດຸທີ່ສະໜອງໃຫ້ໂດຍອີງໃສ່ການປ່ຽນແປງທາງພັນທຸກຳ, ລວມທັງ SNPs, InDels, SVs ແລະ CNVs.ມັນສະຫນອງການວິເຄາະພື້ນຖານທັງຫມົດທີ່ຈໍາເປັນສໍາລັບການອະທິບາຍການປ່ຽນແປງວິວັດທະນາແລະລັກສະນະທາງພັນທຸກໍາຂອງປະຊາກອນ, ເຊັ່ນ: ໂຄງສ້າງປະຊາກອນ, ຄວາມຫຼາກຫຼາຍທາງພັນທຸກໍາ, ຄວາມສໍາພັນຂອງ phylogeny, ແລະອື່ນໆ. ມັນຍັງປະກອບດ້ວຍການສຶກສາກ່ຽວກັບການໄຫຼວຽນຂອງ gene, ເຊິ່ງສ້າງຄວາມເຂັ້ມແຂງໃຫ້ການຄາດຄະເນຂະຫນາດປະຊາກອນທີ່ມີປະສິດທິພາບ, ເວລາຄວາມແຕກຕ່າງ.
-
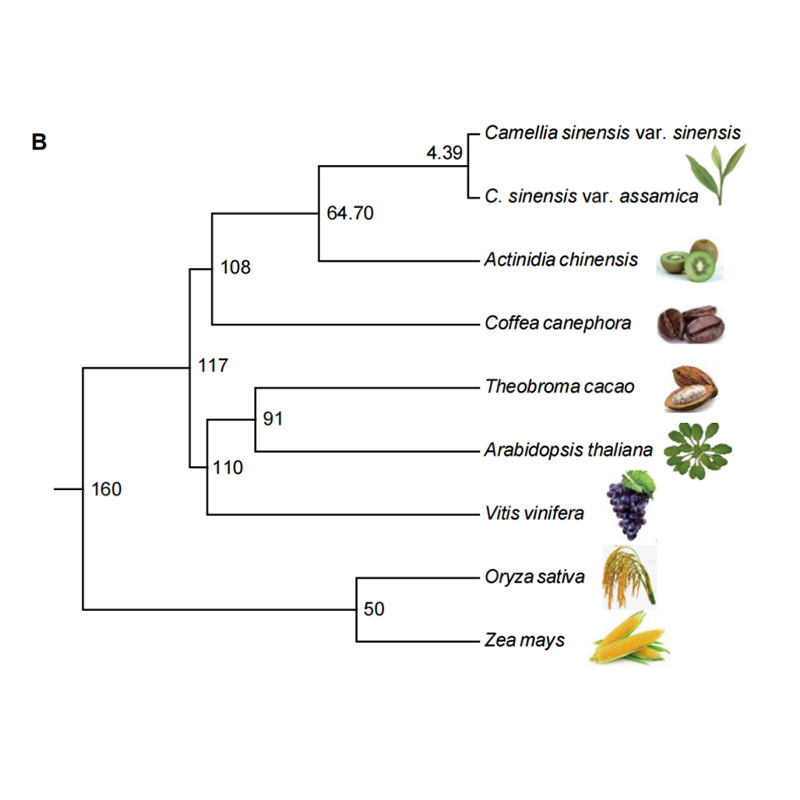
ພັນທຸ ກຳ ປຽບທຽບ
genomics ປຽບທຽບຫມາຍຄວາມວ່າການປຽບທຽບລໍາດັບ genome ທີ່ສົມບູນແລະໂຄງສ້າງຂອງຊະນິດຕ່າງໆ.ລະບຽບວິໄນນີ້ມີຈຸດປະສົງເພື່ອເປີດເຜີຍວິວັດທະນາການຂອງຊະນິດພັນ, ການທໍາງານຂອງພັນທຸກໍາ, ກົນໄກການລະບຽບການ gene ໃນລະດັບ genome ໂດຍການກໍານົດໂຄງສ້າງລໍາດັບແລະອົງປະກອບທີ່ອະນຸລັກຫຼືແຕກຕ່າງກັນໃນທົ່ວຊະນິດຕ່າງໆ.ການສຶກສາ genomics ປຽບທຽບແບບປົກກະຕິປະກອບມີການວິເຄາະໃນຄອບຄົວ gene, ການພັດທະນາວິວັດທະນາການ, ການຊໍ້າຊ້ອນຂອງ genome ທັງຫມົດ, ຄວາມກົດດັນການຄັດເລືອກ, ແລະອື່ນໆ.
-
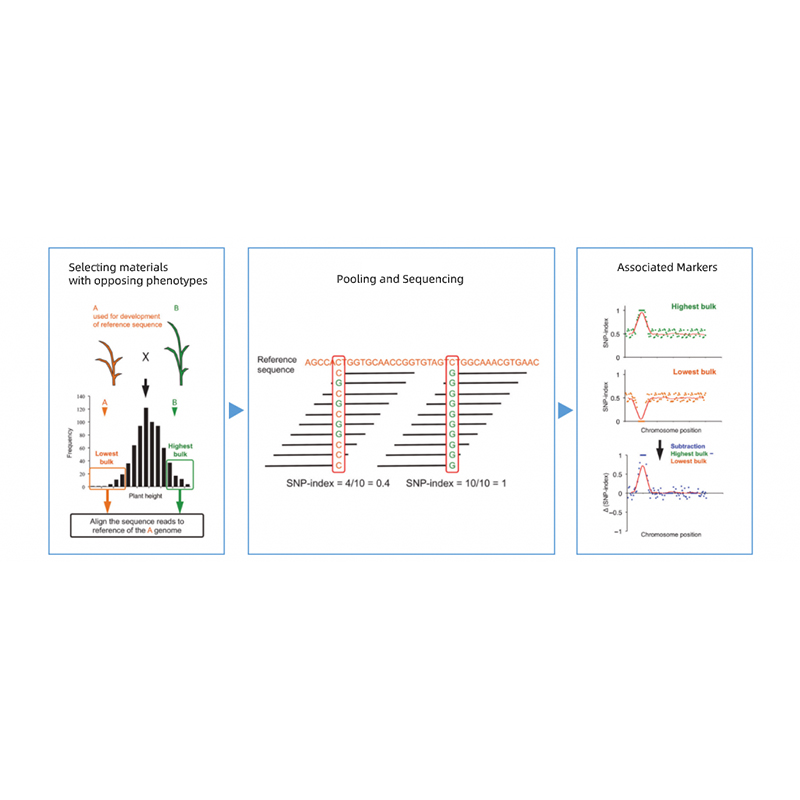
ການວິເຄາະ Segregant ເຕັມ
ການວິເຄາະແບ່ງແຍກເປັນຈຳນວນຫຼວງຫຼາຍ (BSA) ແມ່ນເຕັກນິກທີ່ໃຊ້ເພື່ອກໍານົດຕົວໝາຍພັນທຸກໍາທີ່ກ່ຽວຂ້ອງກັບ phenotype ຢ່າງໄວວາ.ຂະບວນການເຮັດວຽກຕົ້ນຕໍຂອງ BSA ປະກອບມີການເລືອກເອົາສອງກຸ່ມຂອງບຸກຄົນທີ່ມີ phenotypes ກົງກັນຂ້າມທີ່ສຸດ, ລວບລວມ DNA ຂອງບຸກຄົນທັງຫມົດເພື່ອສ້າງເປັນສອງສ່ວນຂອງ DNA, ກໍານົດລໍາດັບຄວາມແຕກຕ່າງລະຫວ່າງສອງສະນຸກເກີ.ເຕັກນິກນີ້ໄດ້ຖືກໃຊ້ຢ່າງກວ້າງຂວາງໃນການກໍານົດເຄື່ອງຫມາຍພັນທຸກໍາທີ່ກ່ຽວຂ້ອງກັບພັນທຸກໍາເປົ້າຫມາຍໃນພັນທຸກໍາຂອງພືດ / ສັດ.





