

High-throughput genotyping, especially on large-scale population, is a fundamental step in genetic association studies, which provides genetic basis for functional gene discovery, evolutionary analysis, etc. Instead of deep whole genome re-sequencing, reduced representation genome sequencing(RRGS) is introduced to minimize sequencing cost per sample, while maintain reasonable efficiency on genetic marker discovery. This is commonly achieved by extracting restriction fragment within given size range, which is named reduced representation library(RRL). Specific-locus amplified fragment sequencing(SLAF-Seq) is a self-developed strategy for de novo SNP discovery and SNP genotyping of large populations.
Technical workflow
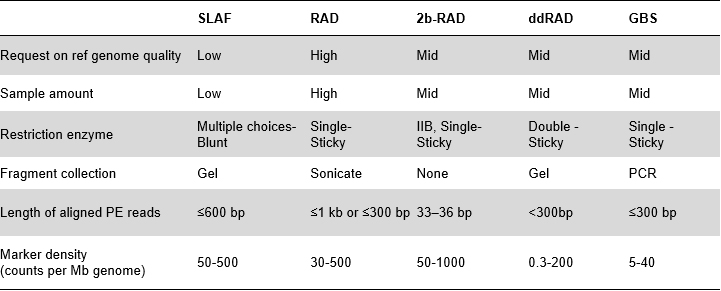
SLAF vs Existing RRL methods
Advantages of SLAF
Higher genetic marker discovery efficiency – Combined with high-throughput sequencing technology, SLAF-Seq could achieve hundreds of thousands of tags discovered within whole genome to fulfill the request of diverse research projects, either with or withour a reference genome.
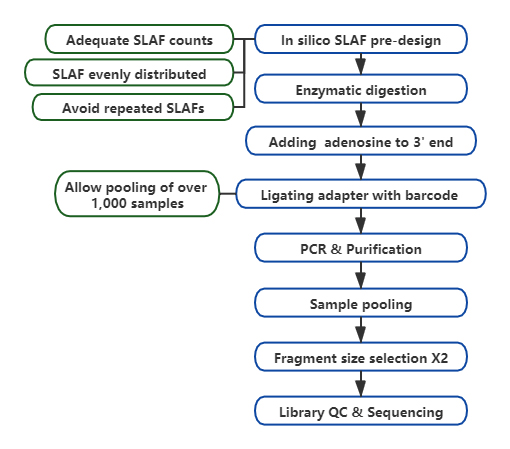
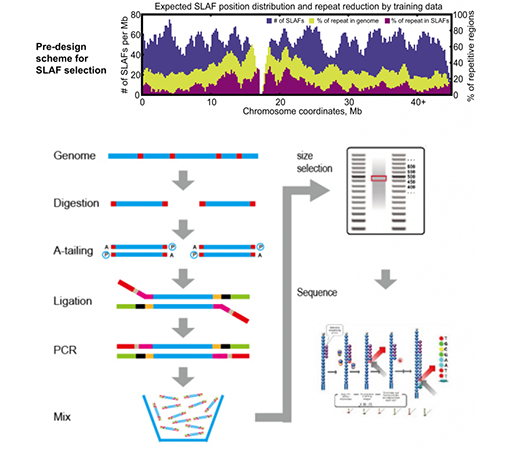
Customized&Flexible experimental design – For different research goal or species, different enzymatic digestion strategies are available including single-enzyme, dual-enzyme and multi-enzyme digestion. Digestion strategy will be pre-evaluated in silico to assure an optimal enzyme design.
High efficiency in enzymatic digestion – Pre-designed enzymatic digestion provides more evenly distributed SLAFs on chromosome. Fragment collection efficient can achieve over 95%.
Avoid repetitive sequence – Percentage of repetitive sequence in SLAF-Seq data is reduced to lower than 5%, especially in species with high level of repetitive elements, such as wheat, maize, etc.
Self-developed bioinformatic workflow – BMK developed an integrated bioinformatic workflow applicable to SLAF-Seq technology to ensure reliability and accuracy of final output.
Application of SLAF
Genetic linkage map
High-density genetic map construction and identification of loci controlling flower-type traits in Chrysanthemum(Chrysanthemum x morifolium Ramat.)
Journal: Horticulture Research Published: 2020.7

GWAS
Identification of a candidate gene associated with isofavone content in soybean seeds using genome-wide association and linkage mapping
Journal: the Plant Journal Published: 2020.08

Evolutionary Genetics
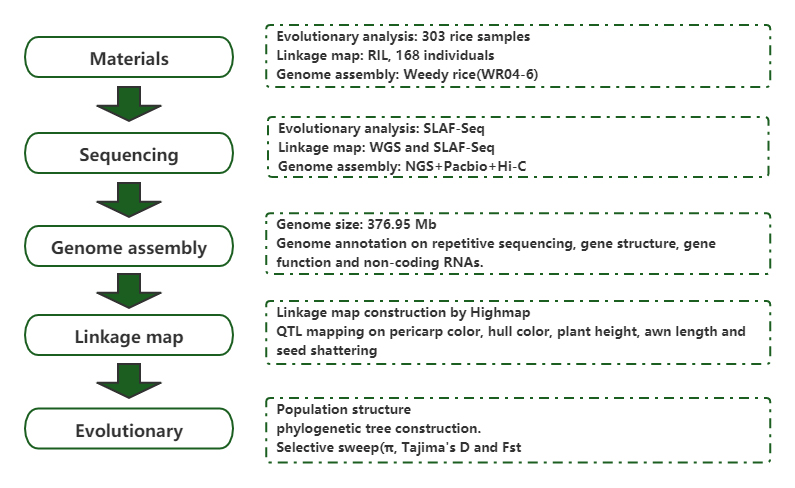
Population genomic analysis and de novo assembly reveal the origin of weedy rice as an evolutionary game
Journal: Molecular Plant Published: 2019.5
Bulked Segregant Analysis (BSA)
GmST1, which encodes a sulfotransferase, confers resistance to soybean mosaic virus strains G2 and G3
Journal: Plant, Cell&Environment Published: 2021.04

Reference
Sun X, Liu D, Zhang X, et al. SLAF-Seq: an efficient method of large-scale de novo SNP discovery and genotyping using high-throughput sequencing[J]. Plos one, 2013, 8(3):e58700
Song X, Xu Y, Gao K, et al. High-density genetic map construction and identification of loci controlling flower-type traits in Chrysanthemum (Chrysanthemum × morifolium Ramat.). Hortic Res. 2020;7:108.
Wu D, Li D, Zhao X, et al. Identification of a candidate gene associated with isoflavone content in soybean seeds using genome-wide association and linkage mapping. Plant J. 2020; 104(4): 950-963.
Sun J, Ma D, Tang L, et al. Population Genomic Analysis and De Novo Assembly Reveal the Origin of Weedy Rice as an Evolutionary Game. Mol Plant. 2019;12(5):632-647. Mol Plant. 2018; 11(11):1360-1376.
Zhao X, Jing Y, Luo Z, et al. GmST1, which encodes a sulfotransferase, confers resistance to soybean mosaic virus strains G2 and G3. Plant Cell Environ. 2021;10.1111/pce.14066
Post time: Jan-04-2022