

Genetyka ewolucyjna








Zalety serwisu
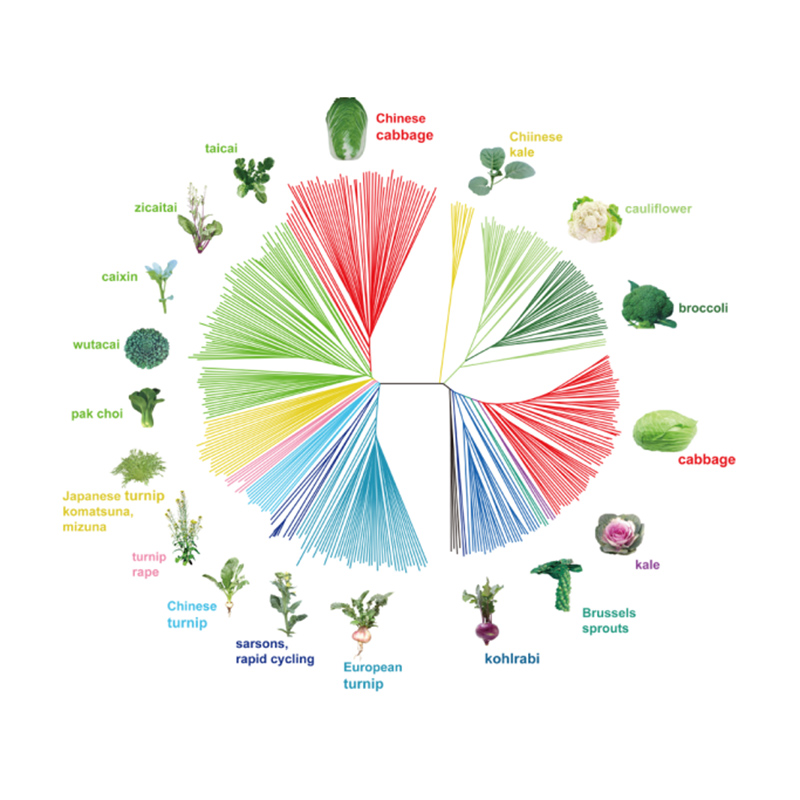
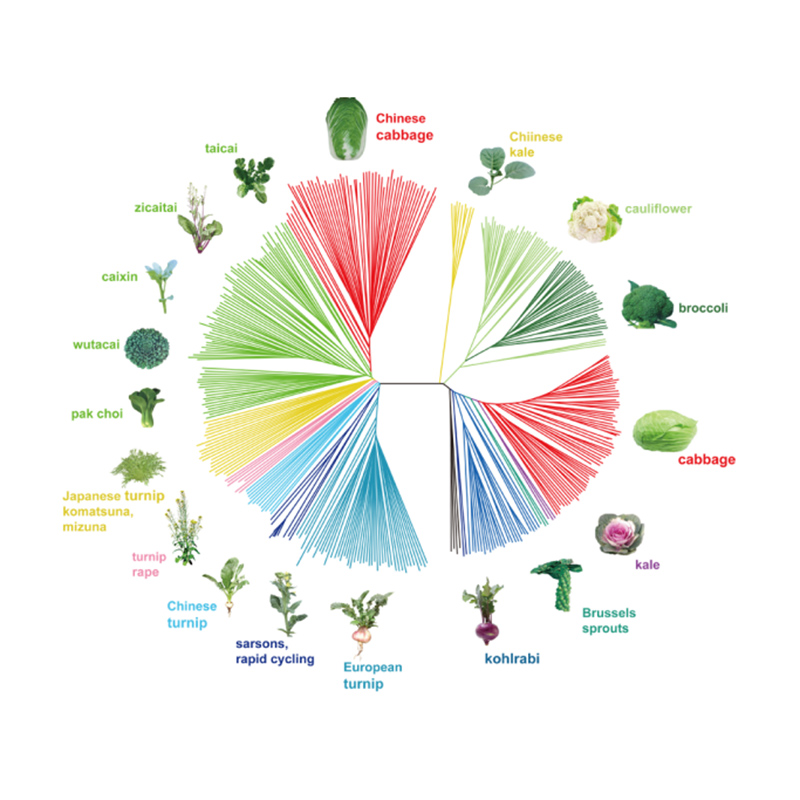
Takagi i in.,Dziennik roślin, 2013
● Szacowanie czasu i szybkości dywergencji gatunków w oparciu o różnice na poziomie nukleotydów i aminokwasów
● Ujawnienie bardziej wiarygodnych powiązań filogenetycznych między gatunkami przy zminimalizowanym wpływie ewolucji zbieżnej i ewolucji równoległej
● Konstruowanie powiązań między zmianami genetycznymi a fenotypami w celu odkrycia genów związanych z cechami
● Szacowanie różnorodności genetycznej, która odzwierciedla potencjał ewolucyjny gatunków
● Szybszy czas realizacji
● Rozległe doświadczenie: BMK zgromadziło ogromne doświadczenie w projektach związanych z populacją i ewolucją przez ponad 12 lat, obejmujących setki gatunków itp. oraz wniosło wkład w ponad 80 projektów wysokiego szczebla opublikowanych w Nature Communications, Molecular Plants, Plant Biotechnology Journal itp.
Specyfikacje usług
Materiały:
Zwykle zaleca się utworzenie co najmniej trzech subpopulacji (np. podgatunków lub szczepów).Każda subpopulacja powinna zawierać nie mniej niż 10 osobników (rośliny > 15, w przypadku rzadkich gatunków można zmniejszyć).
Strategia sekwencjonowania:
* WGS można zastosować w przypadku gatunków z genomem referencyjnym wysokiej jakości, natomiast SLAF-Seq można zastosować w przypadku gatunków z genomem referencyjnym lub bez niego, lub genomu referencyjnego o niskiej jakości.
| Ma zastosowanie do wielkości genomu | WGS | Tagi SLAF (×10 000) |
| ≤ 500 Mb | 10×/osoba | WGS jest bardziej zalecane |
| 500 Mb - 1 Gb | 10 | |
| 1 Gb - 2 Gb | 20 | |
| ≥2 Gb | 30 |
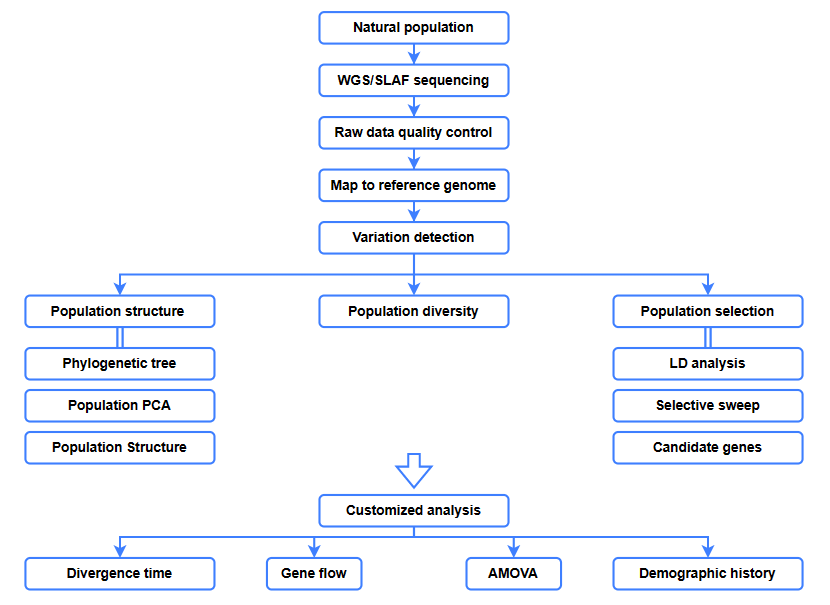
Analizy bioinformatyczne
● Analiza ewolucyjna
● Przemiatanie selektywne
● Przepływ genów
● Historia demograficzna
● Czas rozbieżności
Przykładowe wymagania i dostawa
Przykładowe wymagania:
| Gatunek | Tkanka | WGS-NGS | SLAF |
| Zwierzę
| Tkanka trzewna |
0,5 ~ 1 g
|
0,5 g
|
| Tkanka mięśniowa | |||
| Krew ssaków | 1,5 ml
| 1,5 ml
| |
| Krew drobiowa/rybna | |||
| Zakład
| Świeży Liść | 1~2g | 0,5 ~ 1 g |
| Płatek/łodyga | |||
| Korzeń/Nasiono | |||
| Komórki | Hodowana komórka |
| gDNA | Stężenie | Kwota (ug) | OD260/OD280 |
| SLAF | ≥35 | ≥1,6 | 1,6-2,5 |
| WGS-NGS | ≥1 | ≥0,1 | - |
Przebieg prac serwisowych

Projekt eksperymentu

Dostawa próbek

Budowa biblioteki

Sekwencjonowanie

Analiza danych

Usługi posprzedażowe
*Wszystkie pokazane tutaj wyniki demonstracyjne pochodzą z genomów opublikowanych za pomocą BMKGENE
1.Analiza ewolucji obejmuje konstrukcję drzewa filogenetycznego, struktury populacji i PCA na podstawie zmienności genetycznej.
Drzewo filogenetyczne przedstawia powiązania taksonomiczne i ewolucyjne między gatunkami mającymi wspólnego przodka.
PCA ma na celu wizualizację bliskości między subpopulacjami.
Struktura populacji wskazuje na obecność genetycznie odrębnej subpopulacji pod względem częstotliwości alleli.
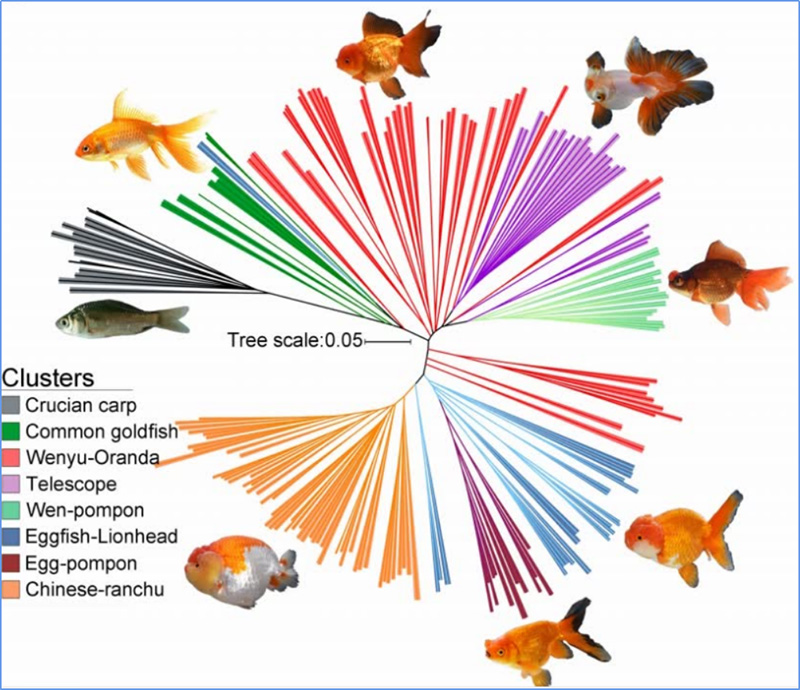
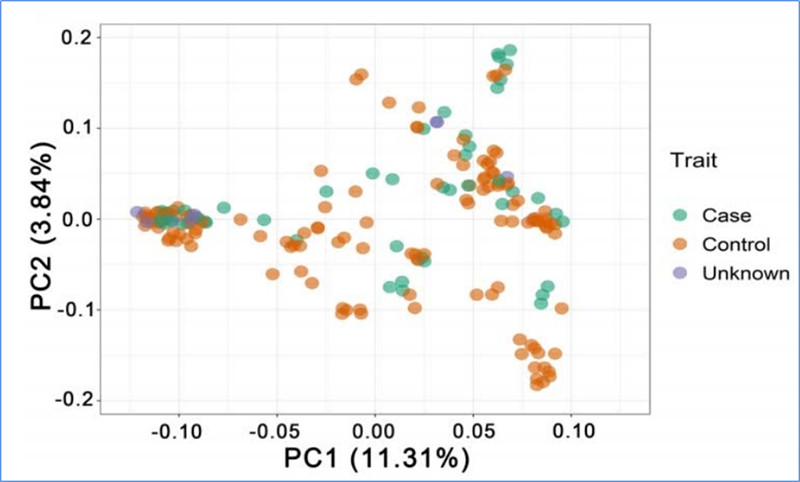
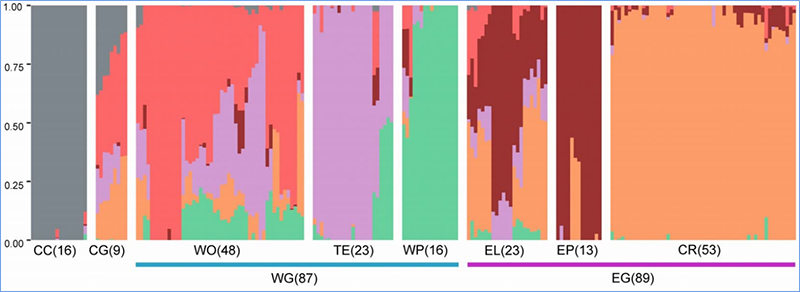
Chena i in.glin.,PNAS, 2020
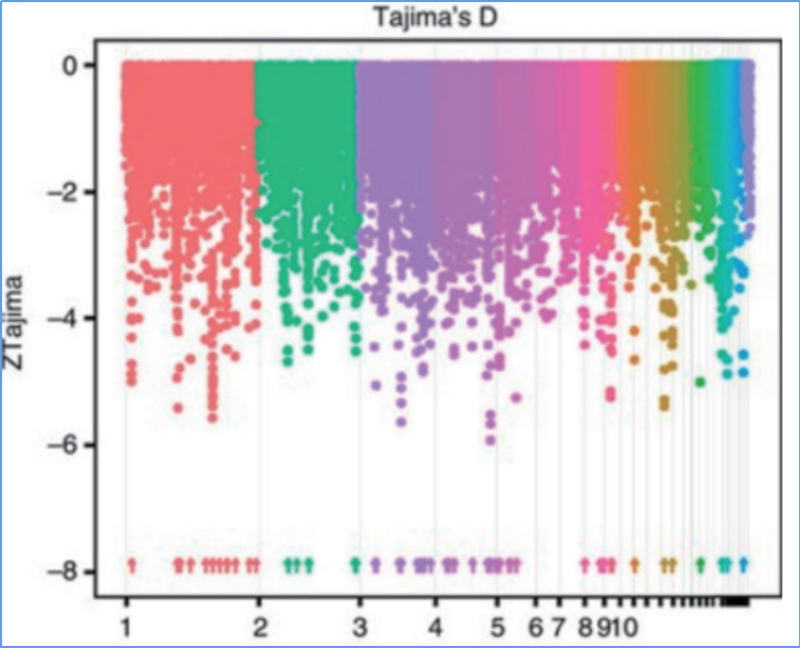
2.Selektywne przemiatanie
Przemiatanie selektywne odnosi się do procesu, w wyniku którego wybierane jest korzystne miejsce i zwiększana jest częstotliwość połączonych miejsc neutralnych, a zmniejszana jest częstotliwość miejsc niepołączonych, co skutkuje zmniejszeniem zasięgu regionalnego.
Wykrywanie całego genomu w selektywnych regionach przemiatania jest przetwarzane poprzez obliczenie indeksu genetycznego populacji (π, Fst, D Tajimy) wszystkich SNP w przesuwanym oknie (100 Kb) w pewnym kroku (10 Kb).
Różnorodność nukleotydów (π)
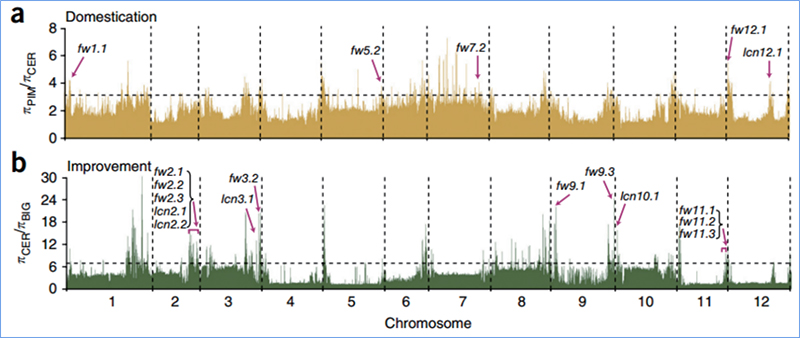
Tajima D
Indeks fiksacji (Fst)
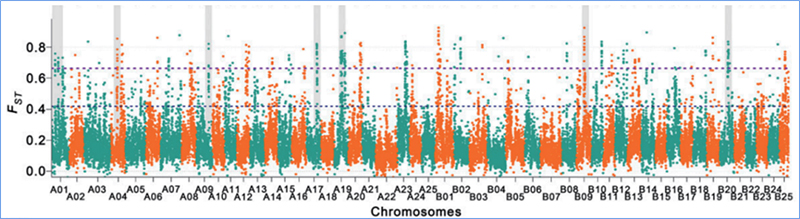
Wu i in.glin.,Roślina molekularna, 2018
3. Przepływ genów
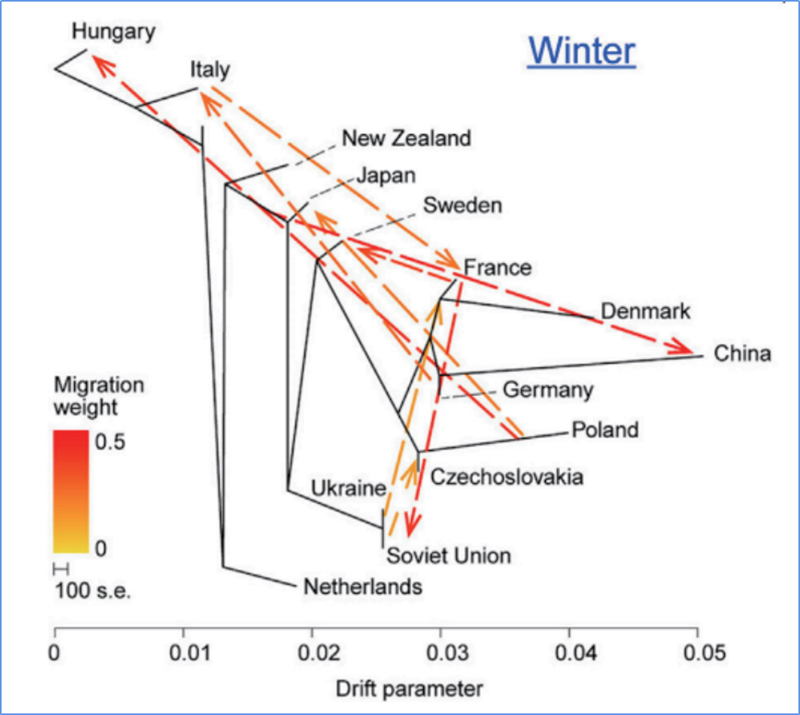
Wu i in.glin.,Roślina molekularna, 2018
4.Historia demograficzna
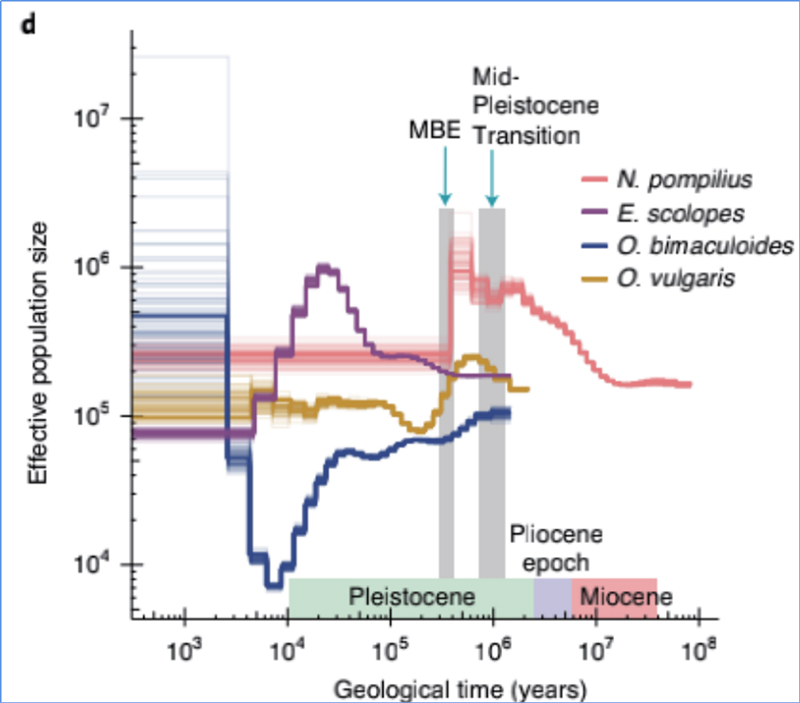
Zhang i in.glin.,Ekologia i ewolucja przyrody, 2021
5.Czas dywergencji
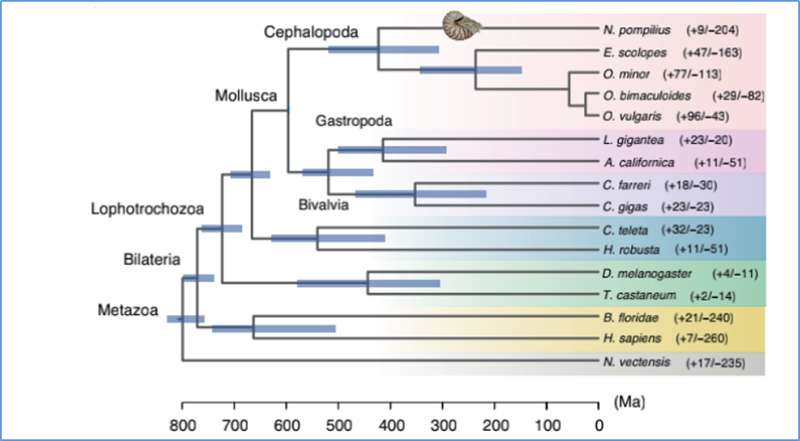
Zhang i in.glin.,Ekologia i ewolucja przyrody, 2021
Sprawa BMK
Mapa zmienności genomu zapewnia wgląd w podstawy genetyczne selekcji jarej kapusty pekińskiej (Brassica rapa ssp. Pekinensis)
Opublikowany: Roślina molekularna, 2018
Strategia sekwencjonowania:
Ponowne sekwencjonowanie: głębokość sekwencjonowania: 10×
Kluczowe wyniki
W tym badaniu 194 kapusty pekińskiej poddano ponownemu sekwencjonowaniu ze średnią głębokością 10×, co dało 1 208 499 SNP i 416 070 InDels.Analiza filogenetyczna tych 194 linii wykazała, że linie te można podzielić na trzy ekotypy: wiosenny, letni i jesienny.Ponadto struktura populacji i analiza PCA wykazały, że wiosenna kapusta pekińska pochodzi z jesiennej kapusty w Shandong w Chinach.Zostały one następnie sprowadzone do Korei i Japonii, skrzyżowane z lokalnymi liniami, a niektóre ich odmiany o późnym pośpiechu zostały sprowadzone z powrotem do Chin i ostatecznie stały się wiosenną kapustą pekińską.
Skanowanie całego genomu jarej kapusty pekińskiej i jesiennej kapusty podczas selekcji ujawniło 23 loci genomowe, które przeszły silną selekcję, z których dwa pokrywały się z regionem kontrolującym czas pośpiechu w oparciu o mapowanie QTL.Stwierdzono, że te dwa regiony zawierają kluczowe geny regulujące kwitnienie, BrVIN3.1 i BrFLC1.Następnie potwierdzono, że te dwa geny są zaangażowane w czas pośpiechu w badaniu transkryptomu i eksperymentach transgenicznych.
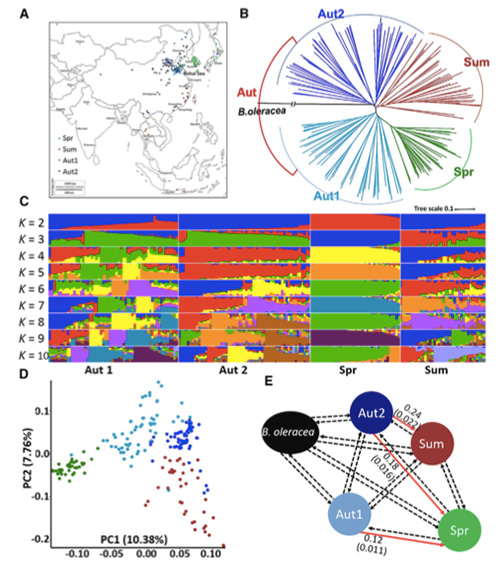 Analiza struktury populacji kapusty pekińskiej | 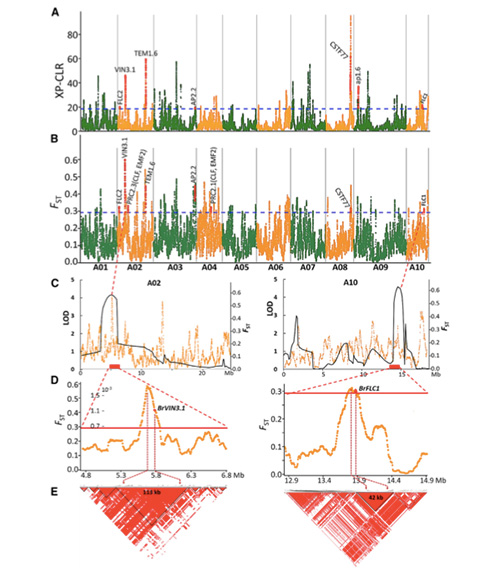 Informacje genetyczne na temat selekcji kapusty pekińskiej |
Tongbing i in.„Mapa zmienności genomu zapewnia wgląd w podstawy genetyczne selekcji jarej kapusty pekińskiej (Brassica rapa ssp.pekinensis)”.Rośliny molekularne,11(2018):1360-1376.





