

TRANSKRYPTOMIA
Natura
KOMUNIKACJA
Charakterystyka transkryptu pełnej długości mutacji SF3B1 w przewlekłej białaczce limfatycznej ujawnia regulację w dół zatrzymanych intronów
Pełne transkrypcje|Sekwencjonowanie nanoporów|Alternatywna analiza izoform
Tło
SPowszechnie donoszono, że mutacje omatyczne w czynniku splicingu SF3B1 są powiązane z różnymi nowotworami, w tym przewlekłą białaczką limfocytową (CLL), czerniakiem błony naczyniowej oka, rakiem piersi itp. Ponadto badania transkryptomiczne o krótkim odczycie ujawniły nieprawidłowe wzorce splicingu indukowane przez mutacje SF3B1.Jednakże badania nad tymi alternatywnymi wzorcami splicingu od dawna ograniczały się do poziomu zdarzenia i braku wiedzy na poziomie izoform ze względu na ograniczenia złożonych transkryptów o krótkim odczycie.W tym przypadku wprowadzono platformę sekwencjonowania nanoporów w celu wygenerowania transkryptów pełnej długości, co umożliwiło badanie izoform AS.
Eksperymentalny projekt
Eksperymenty
Grupowanie:1. CLL-SF3B1(WT) 2. CLL-SF3B1(mutacja K700E);3. Normalne komórki B
Strategia sekwencjonowania:sekwencjonowanie biblioteki MinION 2D, sekwencjonowanie biblioteki PromethION 1D;krótkiego odczytu danych z tych samych próbek
Platforma sekwencjonowania:ONT MinION;ONT PromethION;
Analiza bioinformatyczna
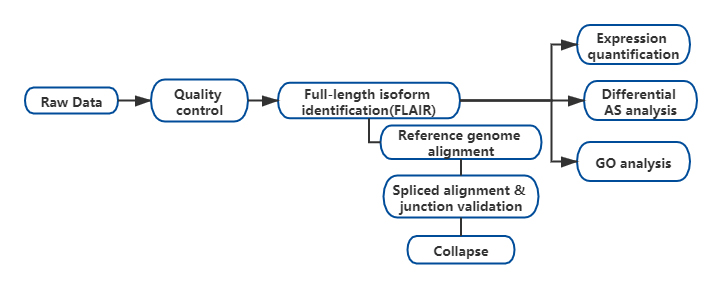
Wyniki
Ałącznie wygenerowano 257 milionów odczytów z 6 próbek CLL i 3 komórek B.Średnio 30,5% tych odczytów zidentyfikowano jako transkrypcje pełnej długości.
FOpracowano analizę alternatywnych izoform RNA (FLAIR) pełnej długości w celu wygenerowania zestawu izoform o wysokim stopniu pewności.FLAIR można podsumować następująco:
Nanopore odczytuje dopasowanie: identyfikuje ogólną strukturę transkryptu w oparciu o genom referencyjny;
Skorekta złącza splicingowego: popraw błędy sekwencji (czerwone) za pomocą miejsca splicingu z intronów z adnotacjami, intronów z danych krótkiego odczytu lub obu;
Collapse: podsumowanie reprezentatywnych izoform w oparciu o łańcuchy połączeń splotowych (zestaw pierwszego przejścia).Wybierz izofrom o wysokiej pewności na podstawie liczby wspierających odczytów (próg: 3).
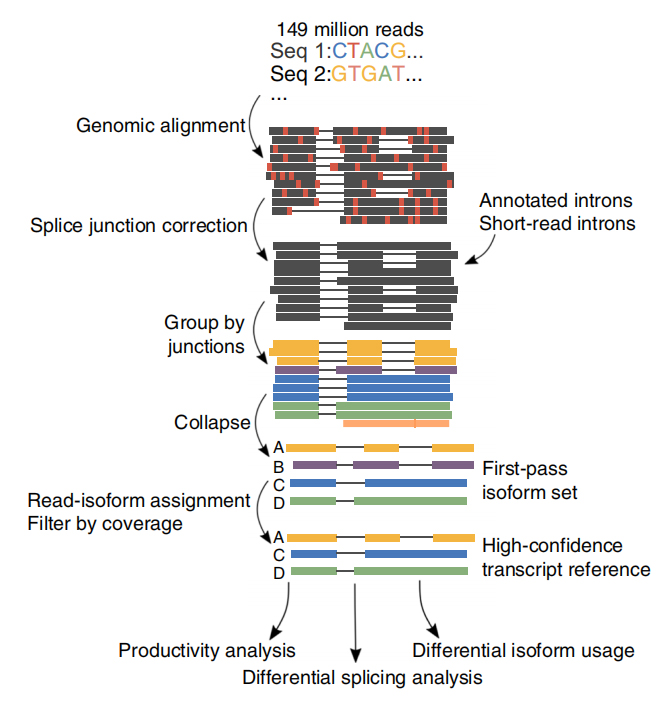
Rycina 1. Analiza FLAIR w celu identyfikacji pełnej długości izoform związanych z mutacją SF3B1 w CLL
FLAIR Zidentyfikowano 326 699 izoform o wysokim stopniu pewności splicingu, z których 90% to nowe izoformy.Stwierdzono, że większość z tych niezanotowanych izoform to nowe kombinacje znanych połączeń splicingowych (142 971), podczas gdy pozostałe nowe izoformy zawierały albo zachowany intron (21 700), albo nowy ekson (3594).
Lsekwencje odczytywane podczas jednorazowego odczytu umożliwiają identyfikację zmutowanych miejsc splicingowych zmienionych w SF3B1-K700E na poziomie izoformy.Stwierdzono, że 35 alternatywnych 3'SS i 10 alternatywnych 5'SS ma znacząco zróżnicowany splicing pomiędzy SF3B1-K700E i SF3B1-WT.33 z 35 zmian zostało nowo odkrytych na podstawie długo czytanych sekwencji.W danych Nanopore rozkład odległości między 3'SS zmienionymi SF3B1-K700E a pikami miejsc kanonicznych wynosi około -20 pz, co znacznie różni się od rozkładu kontrolnego, podobnego do tego, co odnotowano w sekwencjach krótkiego odczytu CLL.Analizowano izoformy genu ERGIC3, przy czym stwierdzono, że w SF3B1-K700E występuje liczniej nowa izoforma zawierająca proksymalne miejsce splicingu.Zarówno bliższy, jak i dalszy 3'SS powiązano z odrębnymi wzorcami AS generującymi wiele izoform.
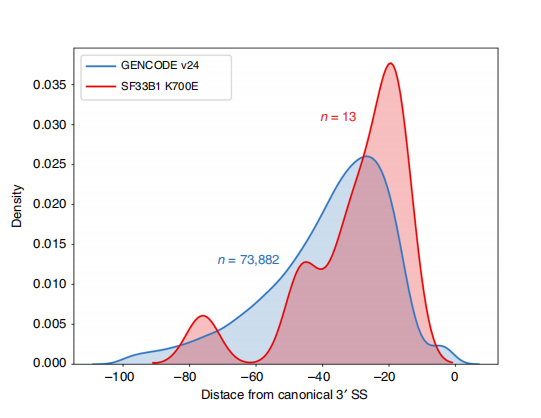
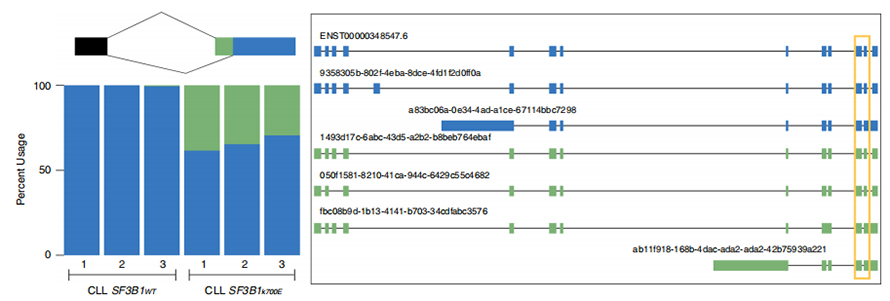
Rysunek 2. Alternatywne wzory splicingu 3′ zidentyfikowane na podstawie danych sekwencjonowania nanoporów
Analiza wykorzystania zdarzeń IR była przez długi czas ograniczona w analizie opartej na krótkim odczycie ze względu na pewność co do identyfikacji i kwantyfikacji IR.Ekspresję izoform IR w SF3B1-K700E i SF3B1-WT określono ilościowo na podstawie sekwencji nanoporów, ujawniając globalną regulację w dół izoform IR w SF3B1-K700E.
Rysunek 4. Intensywność rolnictwa i łączność sieciowa w trzech systemach rolniczych (A i B);Losowa analiza lasów (C) i związek między intensywnością rolnictwa a kolonizacją AMF (D)
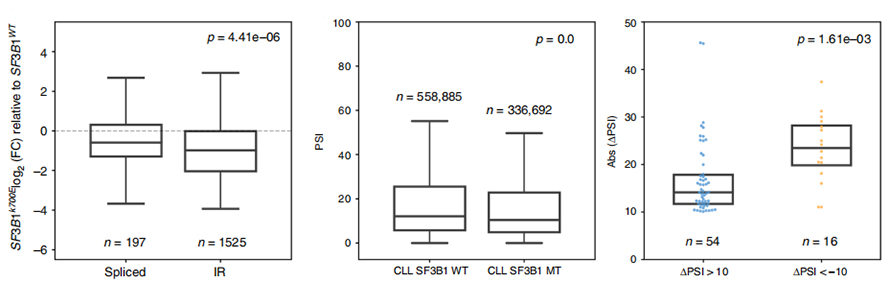
Rysunek 3. Zdarzenia związane z wynajmem intronów są silniej regulowane w dół w CLL SF3B1-K700E
Technologia
Sekwencjonowanie z długim odczytem Nanopore
NSekwencjonowanie anopore to technologia sekwencjonowania sygnału elektrycznego w czasie rzeczywistym pojedynczej cząsteczki.
Ddwuniciowy DNA lub RNA zwiąże się z nanoporowatym białkiem osadzonym w biofilmie i rozwijającym się pod przewodnictwem białka motorycznego.
DNici NA/RNA przechodzą przez białko kanału nanoporowego z określoną szybkością pod wpływem różnicy napięcia.
Mcząsteczki generują różne sygnały elektryczne w zależności od budowy chemicznej.
RWykrywanie sekwencji w czasie rzeczywistym osiąga się poprzez wywołanie bazy.
Wykonanie sekwencjonowania transkryptomu pełnej długości
√ Nasycenie danych
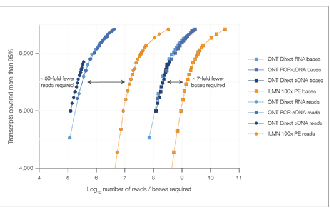
Aby osiągnąć porównywalne nasycenie danymi, potrzeba 7 razy mniej odczytów.
√ Identyfikacja struktury transkrypcji
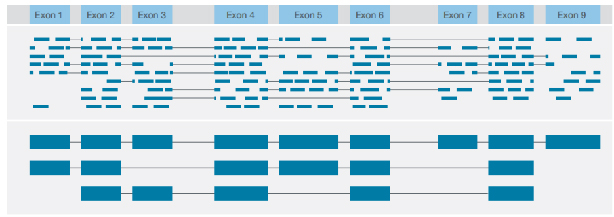
Identyfikacja różnych wariantów strukturalnych z konsensusowym odczytem pełnej długości każdego transkryptu
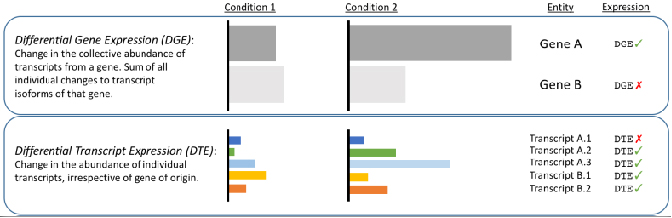
√ Analiza różnicowa na poziomie transkrypcji - odkryj zmiany ukryte przez krótkie odczyty
Odniesienie
Tang AD, Soulette CM, Baren MJV i in.Charakterystyka transkryptu pełnej długości mutacji SF3B1 w przewlekłej białaczce limfatycznej ujawnia regulację w dół zatrzymanych intronów [J].Komunikacja przyrodnicza.
Technologia i najważniejsze informacje ma na celu podzielenie się najnowszymi pomyślnymi zastosowaniami różnych technologii sekwencjonowania o dużej przepustowości w różnych obszarach badawczych, a także genialnymi pomysłami w projektowaniu eksperymentów i eksploracji danych.
Czas publikacji: 08 stycznia 2022 r