

Niereferencyjne sekwencjonowanie mRNA – Illumina








Cechy
● Niezależny od jakiegokolwiek genomu referencyjnego,
● Dane można wykorzystać do analizy struktury i ekspresji transkryptów
● Identyfikuj zmienne miejsca przycinania
Zalety serwisu
● Dostarczanie wyników w oparciu o BMKCloud: Wyniki są dostarczane w postaci pliku danych i interaktywnego raportu za pośrednictwem platformy BMKCloud, która umożliwia przyjazny dla użytkownika odczyt złożonych wyników analiz i dostosowaną do indywidualnych potrzeb eksplorację danych w oparciu o standardową analizę bioinformatyczną.
● Usługi posprzedażowe: Usługi posprzedażowe ważne przez 3 miesiące po zakończeniu projektu, obejmujące monitorowanie projektu, rozwiązywanie problemów, pytania i odpowiedzi dotyczące wyników itp.
Przykładowe wymagania i dostawa
Przykładowe wymagania:
Nukleotydy:
| Stężenie (ng/μl) | Ilość (µg) | Czystość | Uczciwość |
| ≥ 20 | ≥ 0,5 | OD260/280=1,7-2,5 OD260/230=0,5-2,5 Na żelu widoczne jest ograniczone lub żadne zanieczyszczenie białkiem lub DNA. | Dla roślin: RIN≥6,5; Dla zwierząt: RIN≥7,0; 5,0 ≥ 28 S/18 S ≥ 1,0; ograniczone lub żadne wzniesienie linii bazowej |
Tkanka: Waga (sucha): ≥1 g
*W przypadku tkanki o masie mniejszej niż 5 mg zalecamy przesłanie próbki tkanki zamrożonej w ciekłym azocie.
Zawiesina komórek: Liczba komórek = 3 x 1007
*Zalecamy wysyłkę zamrożonego lizatu komórkowego.W przypadku, gdy liczba komórek jest mniejsza niż 5 × 105zaleca się błyskawiczne zamrożenie w ciekłym azocie.
Próbki krwi:
PA×genBloodRNATube;
6 mlLTRIzolu i 2 ml krwi (TRIzol:krew=3:1)
Zalecana dostawa próbek
Pojemnik:
Probówka wirówkowa o pojemności 2 ml (nie zaleca się stosowania folii aluminiowej)
Przykładowe oznakowanie: Grupa+replika, np. A1, A2, A3;B1, B2, B3... ...
Wysyłka:
1.Suchy lód: Próbki należy zapakować do worków i zakopać w suchym lodzie.
2. Probówki RNAstable: Próbki RNA można suszyć w probówkach do stabilizacji RNA (np. RNAstable®) i przesyłać w temperaturze pokojowej.
Przebieg prac serwisowych

Projekt eksperymentu

Dostawa próbek

Ekstrakcja RNA

Budowa biblioteki

Sekwencjonowanie

Analiza danych

Usługi posprzedażowe
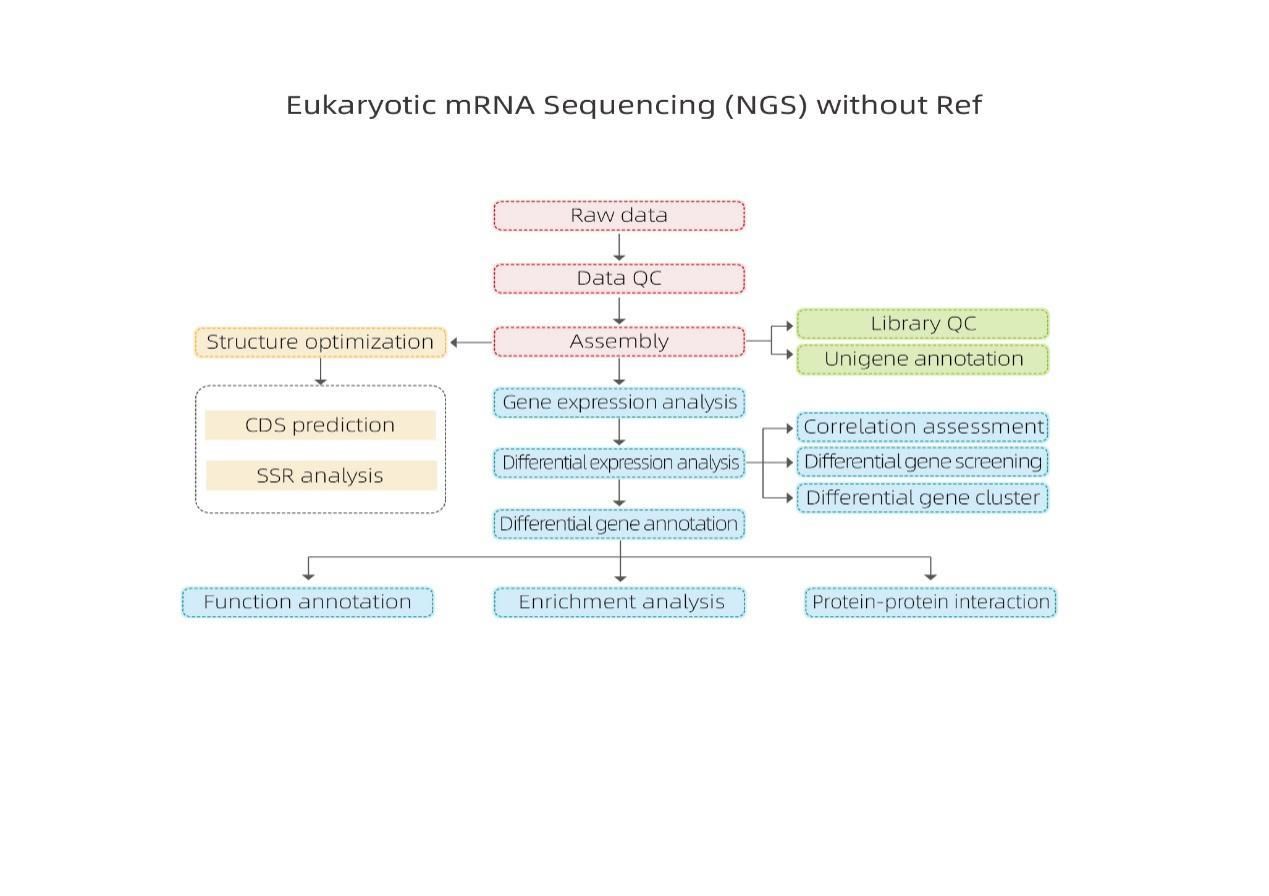
Bioinformatyka
1.mRNA(denovo) Zasada składania
Według Trinity odczyty są podzielone na mniejsze części, zwane K-mer.Te K-mery są następnie wykorzystywane jako nasiona, które można rozszerzyć na kontigi, a następnie składować w oparciu o nakładanie się kontigów.Wreszcie zastosowano tutaj De Bruijna do rozpoznania transkryptów w składnikach.
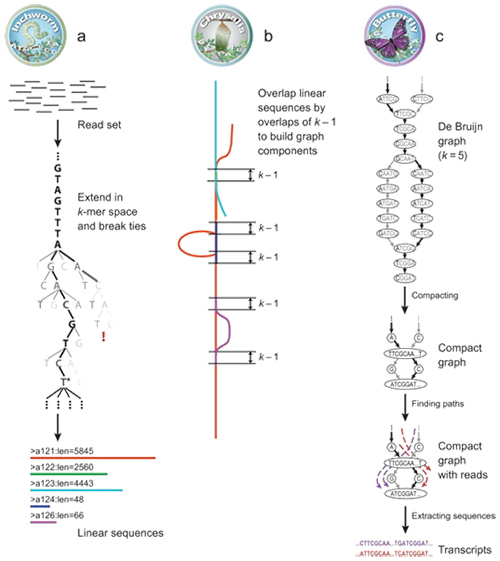
mRNA (De novo) Przegląd Trinity
2.mRNA (De novo) Rozkład poziomu ekspresji genów
RNA-Seq umożliwia bardzo czułą ocenę ekspresji genów.Zwykle wykrywalny zakres ekspresji transkryptów FPKM mieści się w zakresie od 10^-2 do 10^6
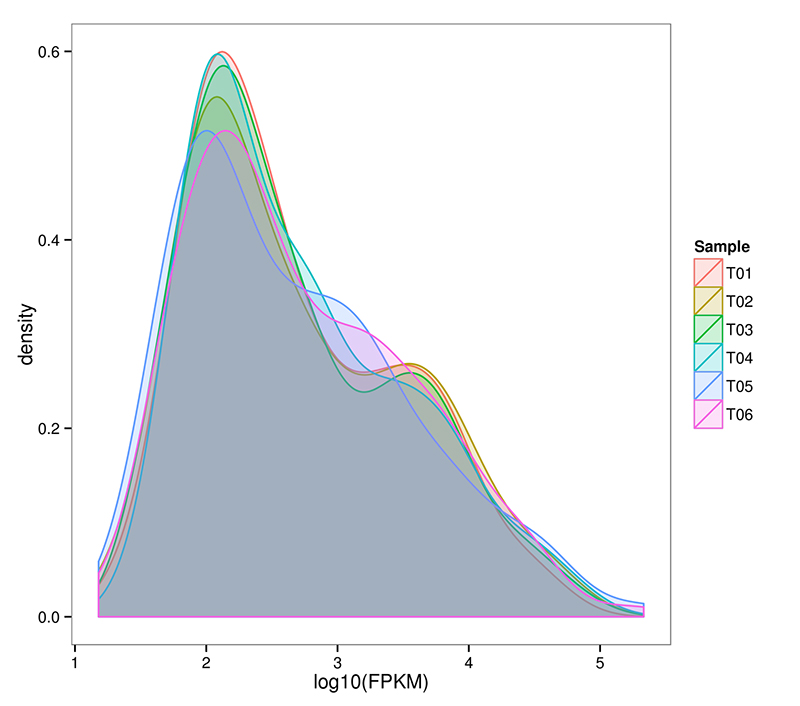
mRNA (De novo) Rozkład gęstości FPKM w każdej próbce
3.MRNA (De novo) Analiza wzbogacania GO w DEG
Baza danych GO (Gene Ontology) to ustrukturyzowany system adnotacji biologicznych zawierający standardowy słownik funkcji genów i produktów genów.Zawiera wiele poziomów, przy czym im niższy poziom, tym bardziej szczegółowe są funkcje.
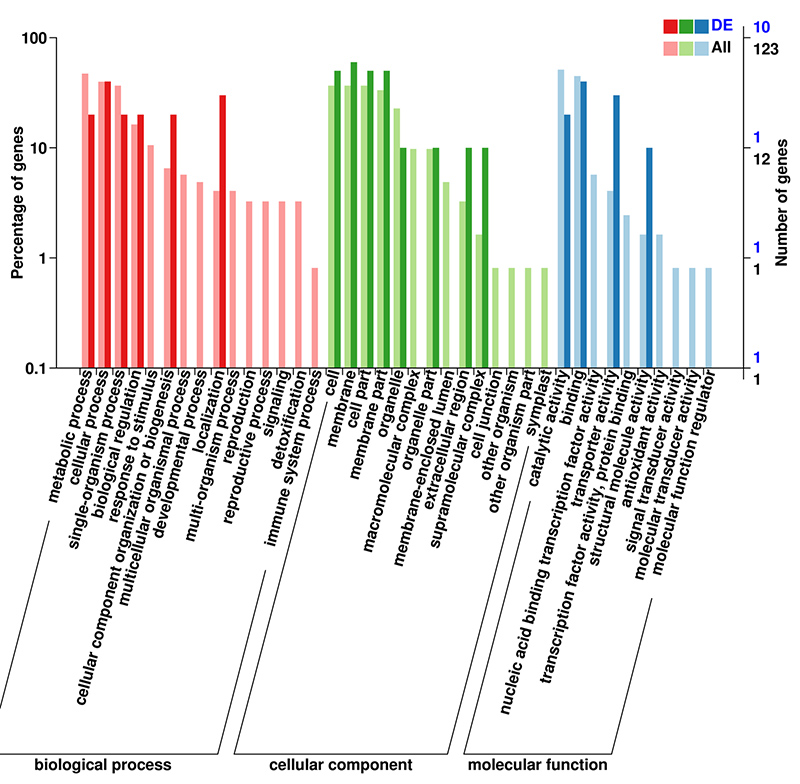
Klasyfikacja mRNA (De novo) GO DEG na drugim poziomie
Sprawa BMK
Analiza transkryptomu metabolizmu sacharozy podczas pęcznienia i rozwoju cebul cebuli (Allium cepa L.)
Opublikowany: granice w nauce o roślinach,2016
Strategia sekwencjonowania
Ilumina HiSeq2500
Zbiór próbek
W badaniach wykorzystano odmianę Utah Yellow Sweet Spain „Y1351”.Liczba zebranych próbek wyniosła
15. dzień po spęcznieniu (DAS) cebulki (średnica 2 cm i masa 3–4 g), 30. DAS (średnica 5 cm i masa 100–110 g) i ~3 w 40. DAS (średnica 7 cm i masa 260–300 gramów).
Kluczowe wyniki
1. na diagramie Venna wykryto łącznie 146 DEG we wszystkich trzech parach stadiów rozwojowych
2. „Transport i metabolizm węglowodanów” był reprezentowany tylko przez 585 unigenów (tj. 7% opisanego COG).
3. Unigenes pomyślnie dodane do bazy danych GO zostały sklasyfikowane w trzech głównych kategoriach dla trzech różnych etapów rozwoju cebul.W kategorii głównej „proces biologiczny” najczęściej reprezentowany był „proces metaboliczny”, a następnie „proces komórkowy”.W głównej kategorii „funkcja molekularna” dwie najczęściej reprezentowane kategorie to „wiązanie” i „aktywność katalityczna”.
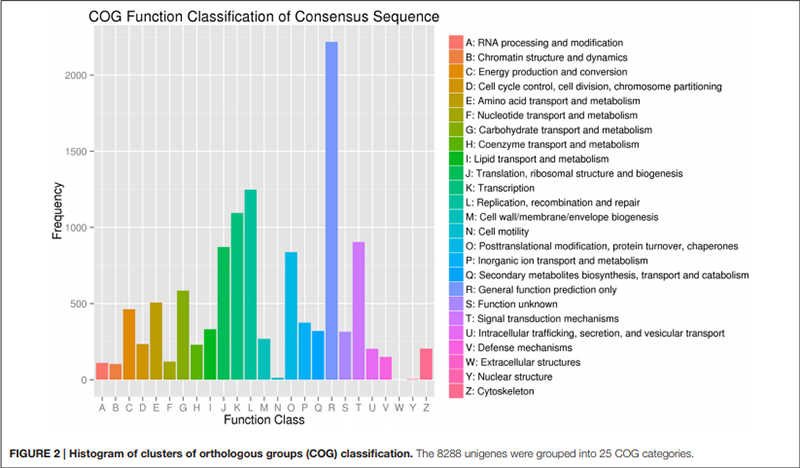 Histogram klasyfikacji klastrów grup ortologicznych (COG). | 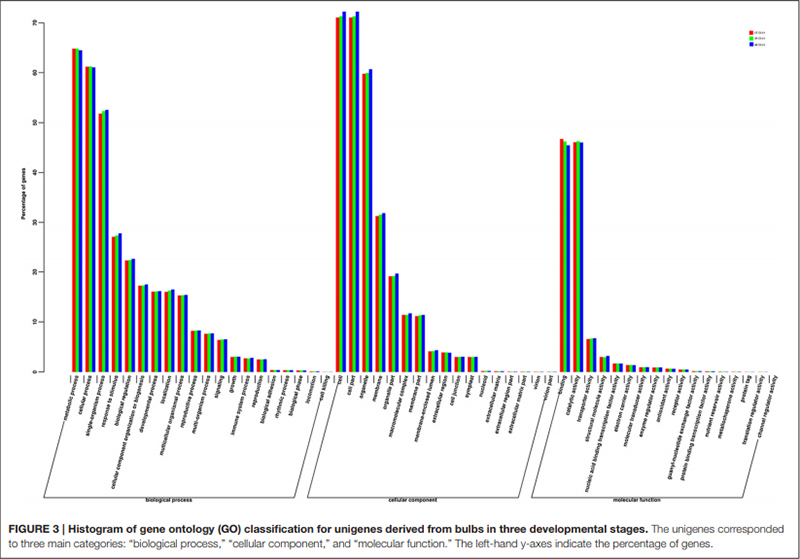 Histogram klasyfikacji ontologii genów (GO) dla unigenów pochodzących z cebul w trzech stadiach rozwojowych |
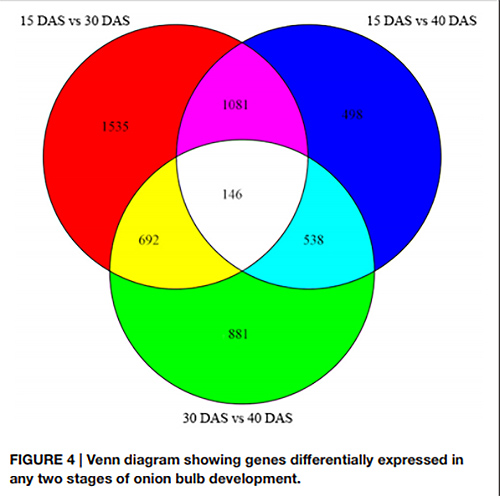 Diagram Venna przedstawiający różną ekspresję genów w dowolnych dwóch stadiach rozwoju cebuli |
Odniesienie
Zhang C, Zhang H, Zhan Z i in.Analiza transkryptomu metabolizmu sacharozy podczas obrzęku i rozwoju cebul cebuli (Allium cepa L.) [J].Frontiers in Plant Science, 2016, 7:1425-.DOI: 10.3389/fpls.2016.01425





