

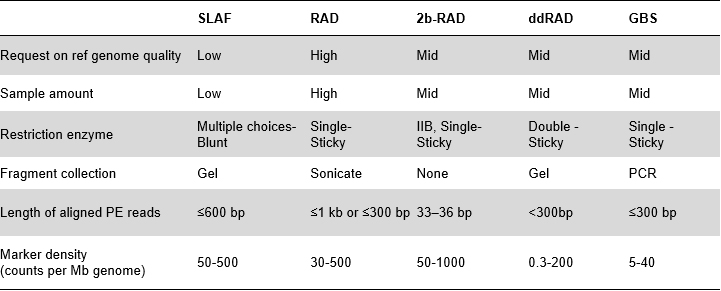
特に大規模集団におけるハイスループットのジェノタイピングは、遺伝的関連研究の基本的なステップであり、機能遺伝子の発見や進化解析などのための遺伝的基礎を提供します。ディープ全ゲノム再配列決定の代わりに、縮小表現ゲノム配列決定(RRGS)が使用されます。 ) は、遺伝子マーカー発見の合理的な効率を維持しながら、サンプルあたりの配列決定コストを最小限に抑えるために導入されています。これは通常、指定されたサイズ範囲内の制限フラグメントを抽出することによって実現され、これは縮小表現ライブラリー (RRL) と呼ばれます。特定遺伝子座増幅フラグメントシークエンシング (SLAF-Seq) は、大集団の de novo SNP 発見と SNP ジェノタイピングのために独自に開発された戦略です。
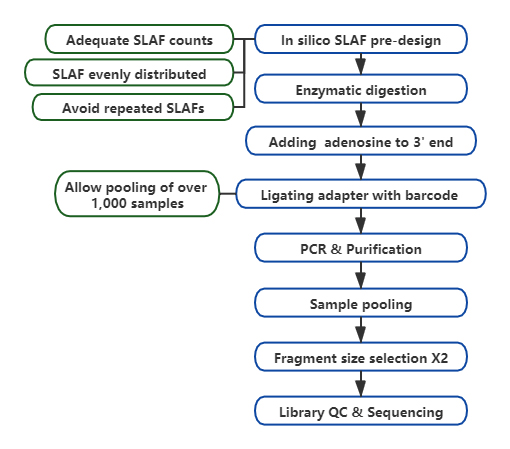
技術的なワークフロー
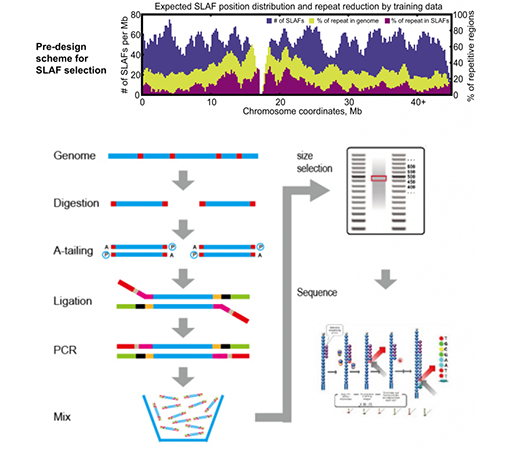
SLAF と既存の RRL 方式の比較
SLAFの利点
より高い遺伝マーカー発見効率– ハイスループットのシーケンス技術と組み合わせることで、SLAF-Seq は全ゲノム内で数十万のタグを発見し、参照ゲノムの有無にかかわらず、多様な研究プロジェクトの要求を満たすことができます。
カスタマイズされた柔軟な実験デザイン– 異なる研究目標や種に対して、単一酵素、二重酵素、多酵素消化などの異なる酵素消化戦略が利用可能です。消化戦略はコンピュータで事前評価され、最適な酵素設計が保証されます。
高い酵素消化効率– 事前に設計された酵素消化により、染色体上でより均一に分散された SLAF が提供されます。フラグメント収集効率は 95% 以上を達成できます。
繰り返しのシーケンスを避ける– SLAF-Seq データ内の反復配列の割合は、特に小麦やトウモロコシなどの反復要素が高レベルにある種では 5% 未満に減少します。
自社開発のバイオインフォマティクスワークフロー– BMK は、最終出力の信頼性と精度を確保するために、SLAF-Seq テクノロジーに適用できる統合バイオインフォマティクス ワークフローを開発しました。
SLAFの適用
遺伝子連鎖地図
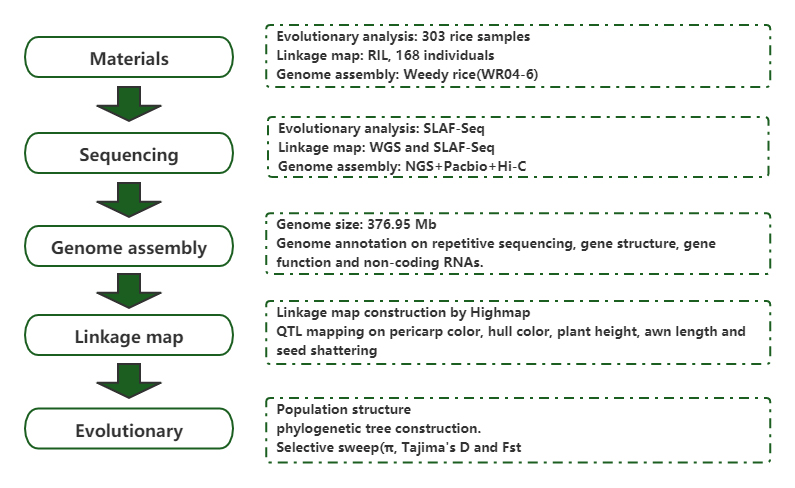
キク(Chrysanthemum x morifolium Ramat.)における高密度遺伝地図の構築と花型形質を制御する遺伝子座の同定
雑誌: 園芸研究 発行: 2020.7



参照
Sun X、Liu D、Zhang X 他SLAF-Seq: ハイスループットシークエンシングを使用した大規模な de novo SNP 発見とジェノタイピングの効率的な方法 [J]。プロスワン、2013、8(3):e58700
Song X、Xu Y、Gao K 他高密度遺伝子地図の構築とキクの花型形質を制御する遺伝子座の同定 (Chrysanthemum × morifolium Ramat.)。ホルティック研究所2020;7:108。
Wu D、Li D、Zhao X 他ゲノムワイドな関連性および連鎖マッピングを使用した、大豆種子中のイソフラボン含有量に関連する候補遺伝子の同定。プラント J. 2020;104(4): 950-963。
Sun J、Ma D、Tang L、他集団ゲノム解析とデノボアセンブリは、進化ゲームとしての雑草イネの起源を明らかにします。モルプラント。2019;12(5):632-647。モルプラント。2018年;11(11):1360-1376。
Zhao X、Jing Y、Luo Z 他硫酸転移酵素をコードするGmST1は、ダイズモザイクウイルスG2株およびG3株に対する耐性を付与します。植物細胞環境。2021;10.1111/pce.14066
投稿時刻: 2022 年 1 月 4 日