

การสร้างจีโนไทป์ที่มีปริมาณงานสูง โดยเฉพาะอย่างยิ่งในประชากรขนาดใหญ่ เป็นขั้นตอนพื้นฐานในการศึกษาความสัมพันธ์ทางพันธุกรรม ซึ่งเป็นพื้นฐานทางพันธุกรรมสำหรับการค้นพบยีนเชิงหน้าที่ การวิเคราะห์วิวัฒนาการ ฯลฯ แทนที่จะจัดลำดับจีโนมใหม่ทั้งหมดในระดับลึก ลดการจัดลำดับจีโนมที่เป็นตัวแทน (RRGS) ) ได้รับการแนะนำเพื่อลดต้นทุนการจัดลำดับต่อตัวอย่าง ขณะเดียวกันก็รักษาประสิทธิภาพที่เหมาะสมในการค้นพบเครื่องหมายทางพันธุกรรมโดยทั่วไปจะทำได้โดยการแตกส่วนข้อจำกัดภายในช่วงขนาดที่กำหนด ซึ่งเรียกว่าไลบรารีการเป็นตัวแทนแบบลดขนาด (RRL)การจัดลำดับแฟรกเมนต์แบบขยายเฉพาะตำแหน่ง (SLAF-Seq) เป็นกลยุทธ์ที่พัฒนาตนเองสำหรับการค้นพบ SNP ใหม่ และจีโนไทป์ SNP ของประชากรจำนวนมาก
ขั้นตอนการทำงานทางเทคนิค
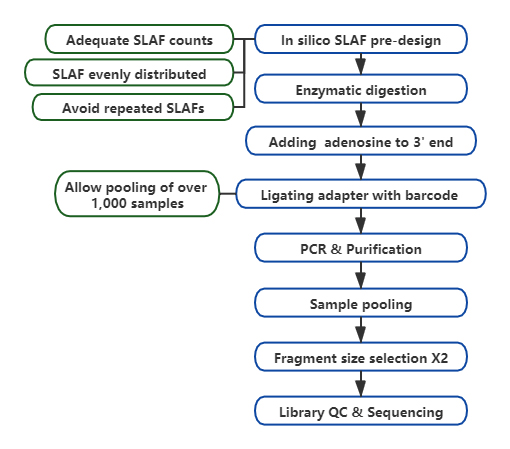
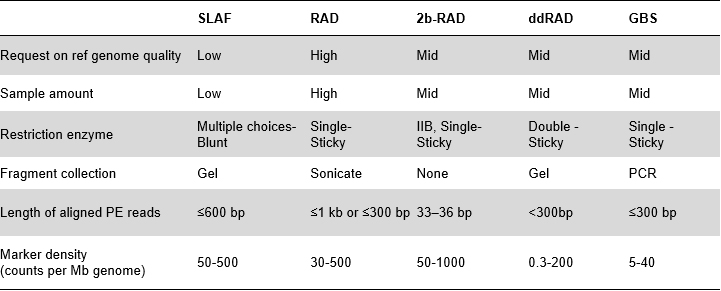
SLAF เทียบกับวิธี RRL ที่มีอยู่
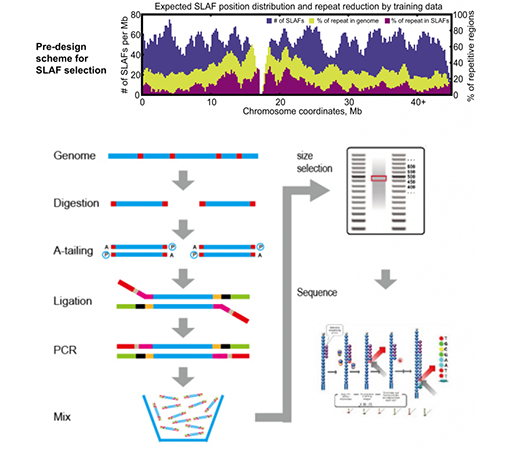
ข้อดีของสลาฟ
ประสิทธิภาพการค้นพบเครื่องหมายทางพันธุกรรมที่สูงขึ้นเมื่อผสมผสานกับเทคโนโลยีการหาลำดับความเร็วสูง SLAF-Seq สามารถบรรลุแท็กนับแสนที่ค้นพบภายในจีโนมทั้งหมด เพื่อตอบสนองคำขอของโครงการวิจัยที่หลากหลาย ไม่ว่าจะมีหรือไม่มีจีโนมอ้างอิงก็ตาม
การออกแบบการทดลองที่ปรับแต่งและยืดหยุ่นได้– สำหรับเป้าหมายการวิจัยหรือสายพันธุ์ที่แตกต่างกัน มีกลยุทธ์การย่อยด้วยเอนไซม์ที่แตกต่างกัน รวมถึงการย่อยด้วยเอนไซม์เดี่ยว เอนไซม์คู่ และการย่อยด้วยเอนไซม์หลายตัวกลยุทธ์การย่อยจะได้รับการประเมินล่วงหน้าในซิลิโกเพื่อให้มั่นใจว่าการออกแบบเอนไซม์ที่เหมาะสมที่สุด
ประสิทธิภาพสูงในการย่อยด้วยเอนไซม์– การย่อยด้วยเอนไซม์ที่ออกแบบไว้ล่วงหน้าช่วยให้ SLAFs บนโครโมโซมมีการกระจายเท่าๆ กันมากขึ้นการรวบรวมชิ้นส่วนที่มีประสิทธิภาพสามารถทำได้มากกว่า 95%
หลีกเลี่ยงการเรียงลำดับซ้ำ– เปอร์เซ็นต์ของลำดับที่ซ้ำกันในข้อมูล SLAF-Seq ลดลงเหลือต่ำกว่า 5% โดยเฉพาะในสายพันธุ์ที่มีองค์ประกอบที่ซ้ำกันในระดับสูง เช่น ข้าวสาลี ข้าวโพดเลี้ยงสัตว์ ฯลฯ
ขั้นตอนการทำงานชีวสารสนเทศที่พัฒนาตนเอง– BMK พัฒนาเวิร์กโฟลว์ชีวสารสนเทศแบบบูรณาการที่ใช้ได้กับเทคโนโลยี SLAF-Seq เพื่อให้มั่นใจในความน่าเชื่อถือและความแม่นยำของผลลัพธ์ขั้นสุดท้าย
การประยุกต์ใช้ SLAF
แผนที่การเชื่อมโยงทางพันธุกรรม
การสร้างแผนที่พันธุกรรมความหนาแน่นสูงและการจำแนกตำแหน่งที่ควบคุมลักษณะดอกไม้ในดอกเบญจมาศ (Chrysanthemum x morifolium Ramat.)
วารสาร: การวิจัยพืชสวน Published: 2020.7

กวาส
การจำแนกยีนที่เกี่ยวข้องกับเนื้อหาไอโซฟาโวนในเมล็ดถั่วเหลืองโดยใช้การเชื่อมโยงทั่วทั้งจีโนมและการทำแผนที่การเชื่อมโยง
วารสาร: วารสารพืช Published: 2020.08

พันธุศาสตร์เชิงวิวัฒนาการ
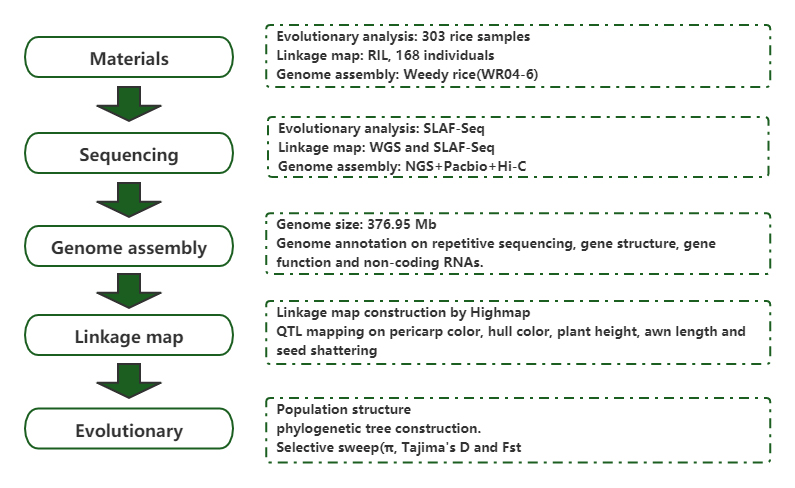
การวิเคราะห์จีโนมประชากรและการประกอบเดอโนโวเผยให้เห็นต้นกำเนิดของข้าววัชพืชในฐานะเกมวิวัฒนาการ
วารสาร: พืชโมเลกุล Published: 2019.5
การวิเคราะห์แยกกลุ่ม (BSA)
GmST1 ซึ่งเข้ารหัสซัลโฟทรานสเฟอเรส ให้ความต้านทานต่อไวรัสโมเสกถั่วเหลืองสายพันธุ์ G2 และ G3
วารสาร: พืช เซลล์&สิ่งแวดล้อม Published: 2021.04

อ้างอิง
ซัน X, Liu D, Zhang X และคณะSLAF-Seq: วิธีการที่มีประสิทธิภาพในการค้นพบ SNP ขนาดใหญ่และการสร้างจีโนไทป์โดยใช้การจัดลำดับความเร็วสูง [J]ประการที่หนึ่ง 2013 8(3):e58700
ซอง X, Xu Y, Gao K และคณะการสร้างแผนที่พันธุกรรมความหนาแน่นสูงและการระบุตำแหน่งที่ควบคุมลักษณะดอกไม้ในเบญจมาศ (เบญจมาศ × morifolium Ramat.)ฮอร์ติกเรส2020;7:108.
Wu D, Li D, Zhao X และคณะการจำแนกยีนที่เกี่ยวข้องกับเนื้อหาไอโซฟลาโวนในเมล็ดถั่วเหลืองโดยใช้การเชื่อมโยงทั่วทั้งจีโนมและการทำแผนที่การเชื่อมโยงโรงงานเจ 2020;104(4): 950-963.
ซัน เจ, มา ดี, ถัง แอล และคณะการวิเคราะห์จีโนมประชากรและการประกอบเดอโนโวเผยให้เห็นต้นกำเนิดของข้าววัชพืชในฐานะเกมวิวัฒนาการโรงงานโมล.2019;12(5):632-647.โรงงานโมล.2561;11(11):1360-1376.
Zhao X, Jing Y, Luo Z และคณะGmST1 ซึ่งเข้ารหัสซัลโฟทรานสเฟอเรส ให้ความต้านทานต่อไวรัสโมเสกถั่วเหลืองสายพันธุ์ G2 และ G3สภาพแวดล้อมของเซลล์พืช2021;10.1111/pce.14066
เวลาโพสต์: Jan-04-2022