

High-throughput genotyping, spesielt på storskala populasjoner, er et grunnleggende trinn i genetisk assosiasjonsstudier, som gir genetisk grunnlag for funksjonell genfunn, evolusjonær analyse, etc. I stedet for dyp helgenom-re-sekvensering, redusert representasjonsgenomsekvensering (RRGS) ) er introdusert for å minimere sekvenseringskostnaden per prøve, samtidig som den opprettholder rimelig effektivitet ved oppdagelse av genetisk markør.Dette oppnås vanligvis ved å trekke ut restriksjonsfragmenter innenfor gitt størrelsesområde, som kalles redusert representasjonsbibliotek (RRL).Spesifikk-locus amplifisert fragmentsekvensering (SLAF-Seq) er en egenutviklet strategi for de novo SNP-oppdagelse og SNP-genotyping av store populasjoner.
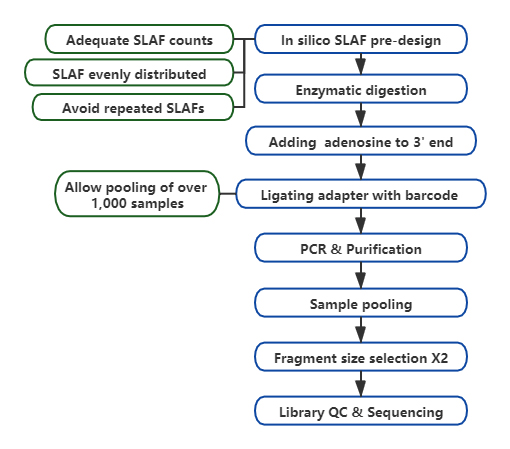
Teknisk arbeidsflyt
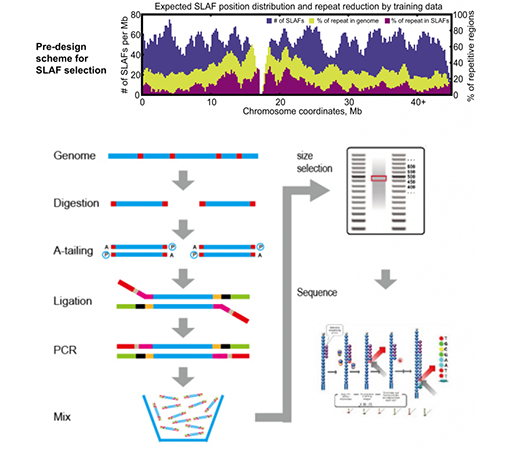
SLAF vs eksisterende RRL-metoder
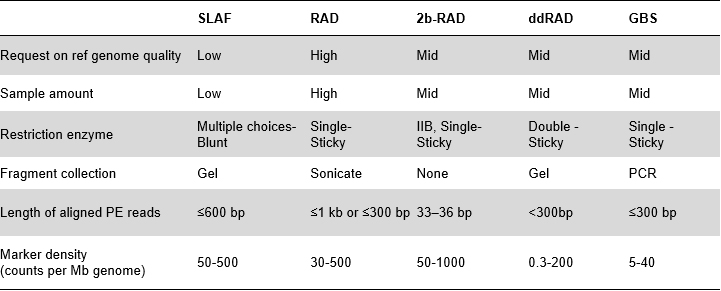
Fordeler med SLAF
Høyere effektivitet for oppdagelse av genetiske markører– Kombinert med high-throughput sekvenseringsteknologi, kan SLAF-Seq oppnå hundretusenvis av tags oppdaget innenfor hele genomet for å oppfylle forespørselen fra ulike forskningsprosjekter, enten med eller uten et referansegenom.
Tilpasset og fleksibel eksperimentell design– For ulike forskningsmål eller arter, er forskjellige enzymatiske fordøyelsesstrategier tilgjengelige, inkludert enkelt-enzym, dual-enzym og multi-enzym fordøyelse.Fordøyelsesstrategi vil bli forhåndsevaluert i silico for å sikre et optimalt enzymdesign.
Høy effektivitet i enzymatisk fordøyelse– Forhåndsdesignet enzymatisk fordøyelse gir mer jevnt fordelte SLAF-er på kromosomet.Fragmentoppsamlingseffektiv kan oppnå over 95 %.
Unngå repeterende sekvenser– Prosentandelen av repeterende sekvens i SLAF-Seq-data er redusert til lavere enn 5 %, spesielt i arter med høyt nivå av repeterende elementer, som hvete, mais, etc.
Egenutviklet bioinformatisk arbeidsflyt– BMK utviklet en integrert bioinformatisk arbeidsflyt som gjelder SLAF-Seq-teknologi for å sikre pålitelighet og nøyaktighet av sluttresultatet.
Anvendelse av SLAF
Genetisk koblingskart
Konstruksjon av genetisk kart med høy tetthet og identifikasjon av loci som kontrollerer trekk av blomstertypen i Chrysanthemum (Chrysanthemum x morifolium Ramat.)
Tidsskrift: Horticulture Research Publisert: 2020.7

GWAS
Identifikasjon av et kandidatgen assosiert med isofavoninnhold i soyafrø ved bruk av genomomfattende assosiasjon og koblingskartlegging
Journal: Plant Journal Publisert: 2020.08

Evolusjonær genetikk
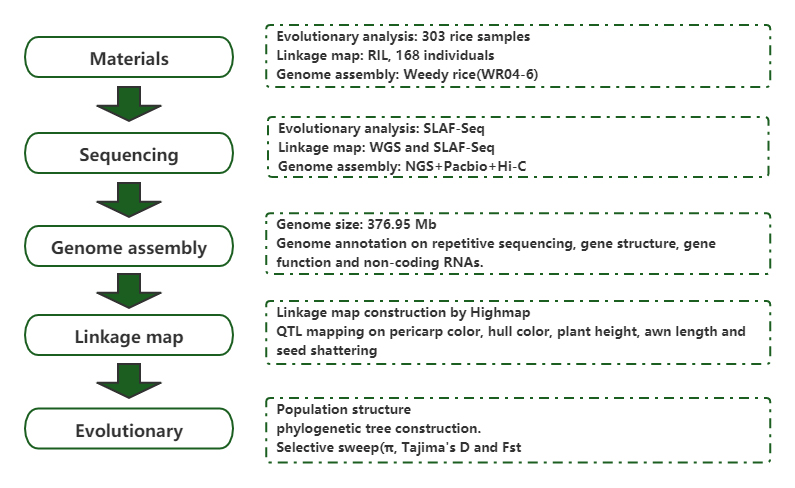
Befolkningsgenomisk analyse og de novo-samling avslører opprinnelsen til ugresset ris som et evolusjonært spill
Tidsskrift: Molecular Plant Publisert: 2019.5
Bulked Segregant Analysis (BSA)
GmST1, som koder for en sulfotransferase, gir resistens mot soyabønnemosaikkvirusstammer G2 og G3
Tidsskrift: Plant, Cell&Environment Publisert: 2021.04

Henvisning
Sun X, Liu D, Zhang X, et al.SLAF-Seq: en effektiv metode for storskala de novo SNP-oppdagelse og genotyping ved bruk av high-throughput-sekvensering [J].Plos en, 2013, 8(3):e58700
Song X, Xu Y, Gao K, et al.Konstruksjon av genetisk kart med høy tetthet og identifisering av loki som kontrollerer trekk av blomstertypen i Chrysanthemum (Chrysanthemum × morifolium Ramat.).Hortic Res.2020;7:108.
Wu D, Li D, Zhao X, et al.Identifikasjon av et kandidatgen assosiert med isoflavoninnhold i soyafrø ved bruk av genomomfattende assosiasjon og koblingskartlegging.Plant J. 2020;104(4): 950-963.
Sun J, Ma D, Tang L, et al.Populasjonsgenomisk analyse og De Novo Assembly avslører opprinnelsen til Weedy Rice som et evolusjonært spill.Mol Plant.2019;12(5):632-647.Mol Plant.2018;11(11):1360-1376.
Zhao X, Jing Y, Luo Z, et al.GmST1, som koder for en sulfotransferase, gir resistens mot soyabønnemosaikkvirusstammer G2 og G3.Plantecellemiljø.2021;10.1111/stk.14066
Innleggstid: Jan-04-2022