

GENOMEVOLUSJON
naturgenetikk
En genomsamling av høy kvalitet fremhever ruggenomiske egenskaper og agronomisk viktige gener
PacBio |Illumina |Bionano optisk kart |Hi-C Genome Assembly |Genetisk kart |Selektive sveiper |RNA-Seq |ISO-seq |SLAF-seq
Biomarker Technologies ga teknisk støtte på Pacbio-sekvensering, Hi-C-sekvensering og dataanalyse i denne studien.
Høydepunkter
1. Det første Rye-genomet av høy kvalitet på kromosomnivå ble oppnådd, som har en enkelt kromosomstørrelse større enn 1 Gb.
2. Sammenlignet med Tu-, Aet- og Hv-genomet ble en unik nylig LTR-RT-hendelse observert i Rye-genomet, som var ansvarlig for utvidelsen av ruggenomets størrelse.
3. Divergensen mellom rug og diploid hvete fant sted etter separasjonen av bygg fra hvete, med divergenstidene for de to hendelsene på omtrent 9,6 og 15 MYA.
Fosforylering av FT-gener kan kontrollere den tidlige overskriften i rug.
4.Selektiv sveipeanalyse indikerer mulig involvering av ScID1 i reguleringen av overskriftsdato og dens sannsynlige valg ved domestisering i rug
Bakgrunn
Bakgrunn
Rug er en verdifull mat- og fôravling, en viktig genetisk ressurs for forbedring av hvete og triticale, og et uunnværlig materiale for effektive komparative genomiske studier i gress.Weining rug, en tidlig blomstrende variant som dyrkes i Kina, er enestående på grunn av sin bredspektrede motstand mot både pulveraktig mugg og striperust.For å forstå det genetiske og molekylære grunnlaget for rugelitegenskaper og for å fremme genomiske studier og avlsstudier i rug og relaterte avlinger, sekvenserte og analyserte vi her genomet til Weining rug.
Prestasjoner
Rye Genom
Rye-genomet ble konstruert ved å kjemme PacBio SMRT-lesninger, kortlest Illumina-sekvensering, så vel som de fra kromatinkonformasjonsfangst (Hi-C), genetisk kartlegging og BioNano-analyse.De sammensatte contigs (7,74 Gb) utgjorde 98,47% av den estimerte genomstørrelsen (7,86 Gb), med 93,67% av contigs (7,25 Gb) tildelt syv kromosomer.Repeterende elementer utgjorde 90,31 % av det samlede genomet.
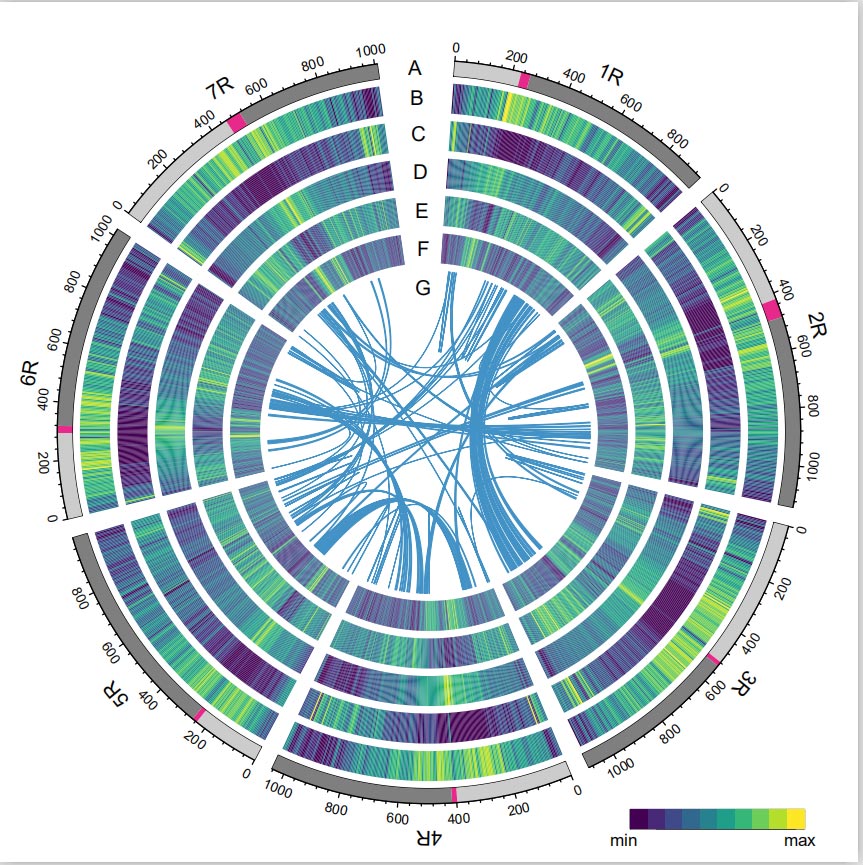
Rye Genom
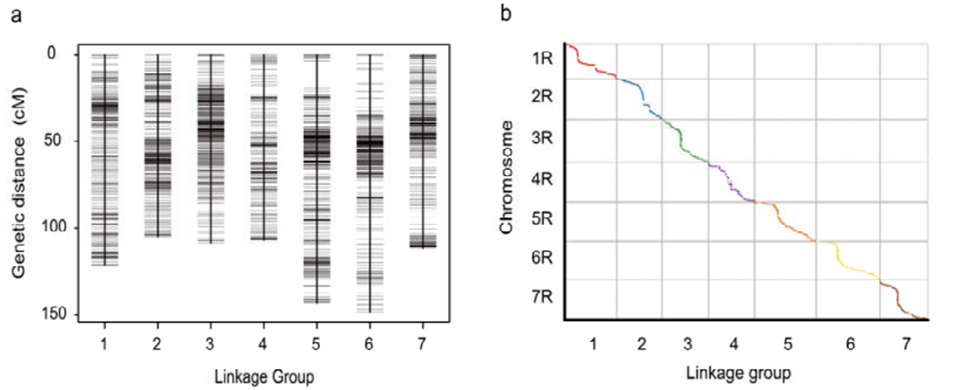
Genetisk koblingskart (WJ) utviklet ved bruk av 295 F2-planter avledet fra kryssing av to ruglandraser (Weining × Jingzhou)
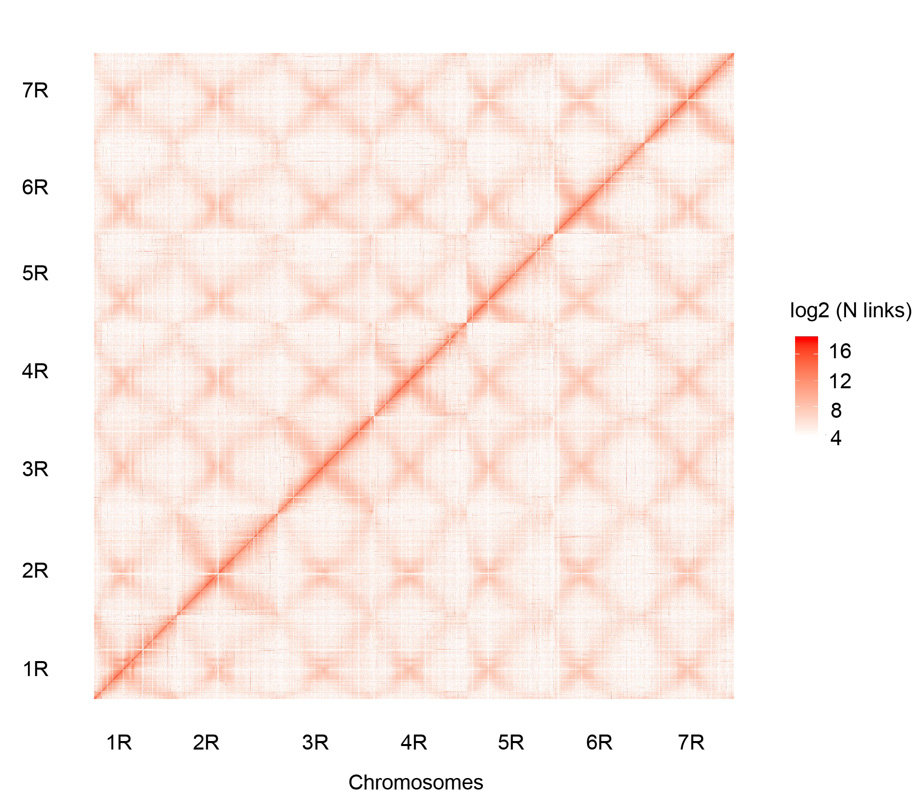
Hi-C kontaktkart over de syv sammensatte Weining rugkromosomene (1R – 7R)
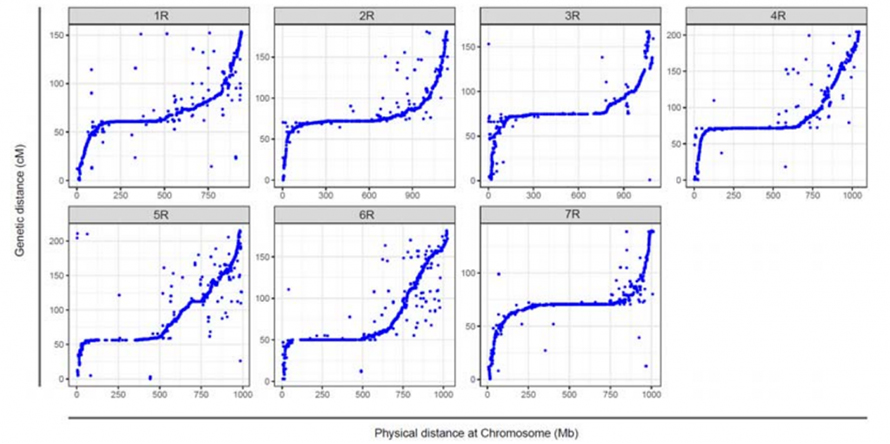
Justering mellom de syv sammensatte kromosomene til Weining rug og de syv rugkoblingsgruppene utviklet ved bruk av Lo7 x Lo255 RIL-populasjon
LTR Assembly Index (LAI)-verdien til ruggenomet ble funnet å være 18,42 og 1393 (96,74%) av de 1440 svært konserverte BUSCO-genene ble identifisert ut. Disse resultatene tyder på at Weining-ruggenomsekvensen er av høy kvalitet i både intergeniske og geniske regioner.Totalt 86.991 proteinkodende gener, inkludert 45.596 gener med høy konfidens (HC) og 41.395 gener med lav konfidens (LC) ble forutsagt.
2. Analyse av TE-er
Analyse av TEer.Totalt 6,99 Gb, som representerer 90,31% av Weining-forsamlingen, ble anmerket som TE-er, som inkluderte 2 671 941 elementer som tilhørte 537 familier.Dette TE-innholdet var klart høyere enn det tidligere rapportert for Ta (84,70%), Tu (81,42%), Aet (84,40%), WEW (82,20%) eller Hv (80,80%).De lange terminale repeterende retrotransposonene (LTR-RTs), inkludert Gypsy, Copia og uklassifiserte RT-elementer, var de dominerende TE-ene, og 1 opptok 84,49 % av annotert TE-innhold og 76,29 % av det sammensatte Weining-genomet;CACTA DNA-transposoner var de nest mest tallrike TE-ene, og utgjorde 11,68 % av annotert TE-innhold og 10,55 % av det sammensatte Weining-genomet.
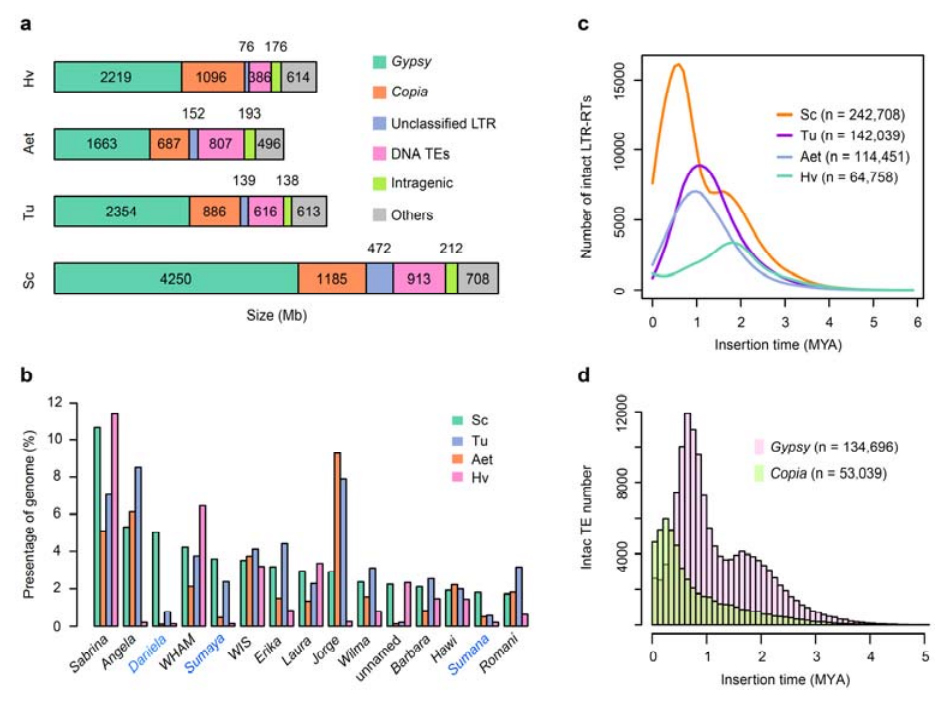
Analyse av transposonelementer av rug
Weining rug hadde en relativt høy andel av nylige innsettinger av LTR-RTer med toppen av amplifikasjonen dukket opp for rundt 0,5 millioner år siden (MYA), som var den nyeste blant de fire artene;den andre toppen, skjedde ca. 1,7 MYA, var eldre og også sett i bygg.På superfamilienivå ble det funnet helt nye utbrudd av Copia-elementer i Weining-rug ved 0,3 MYA, mens forsterkningene av Gypsy RT-er dominerende formet det bimodale distribusjonsmønsteret til LTR-RT-utbruddsdynamikk.
3. Undersøkelse av ruggenomevolusjon og kromosomsyntenier
Divergensen mellom rug og diploid hvete fant sted etter separasjonen av bygg fra hvete, med divergenstidene for de to hendelsene på henholdsvis ca. 9,6 og 15 MYA.1R, 2R, 3R var helt kollineære med henholdsvis gruppe 1, 2 og 3 kromosomer av hvete.4R, 5R, 6R, 7R ble funnet at det finnes storskala fusjoner og segmenter.
4. Analyse av genduplikasjoner og deres innvirkning på stivelsesbiosyntesegener
Spesielt var antallet tandemly dupliserte gener (TDG) og proksimalt dupliserte gener (PDG) av Weining rug begge høyere enn de som ble funnet for Tu, Aet, Hv, Bd og Os.Transponerte dupliserte gener (TrDGs) var også flere enn de spesifikt funnet for Tu og Aet.Ruggenomutvidelse er ledsaget av høyere antall genduplikasjoner.De økte TE-utbruddene i rug kan ha ført til et forhøyet antall TrDGs.
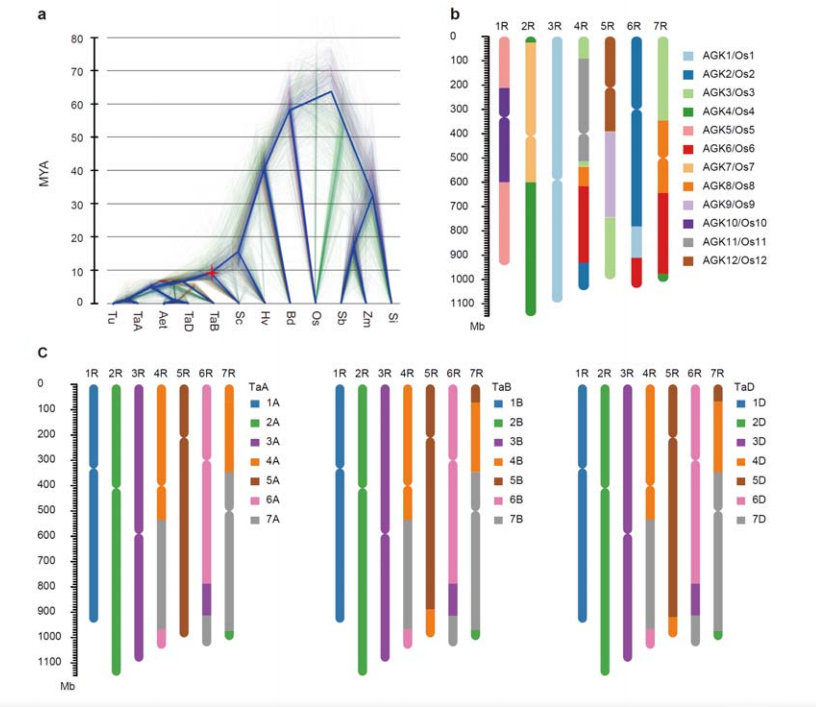
Evolusjons- og kromosomsyntenyanalyser av ruggenom
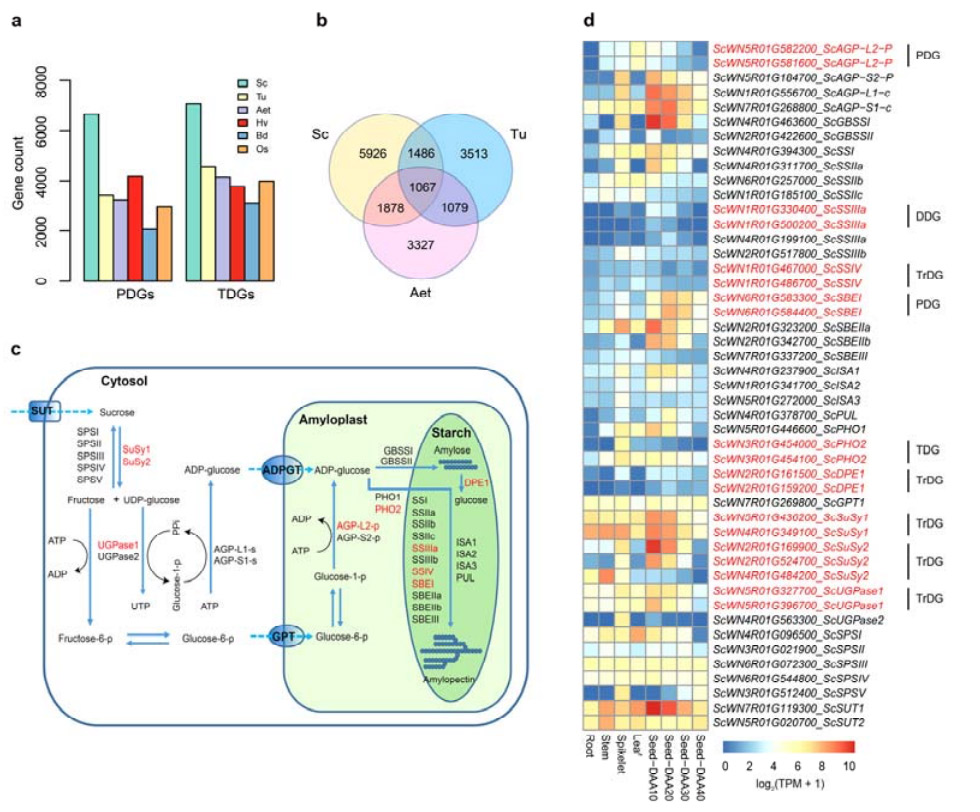
Analyse av ruggenduplikasjoner og deres innvirkning på mangfoldet av stivelsesbiosynteserelaterte gener (SBRG)
5. Disseksjon av rugfrølagringsprotein (SSP) genloci
Fire kromosomale loci (Sec-1 til Sec-4) som spesifiserer rug-SSP-er er identifisert på 1R eller 2R.α-gliadin gener ble utviklet først nylig i hvete og nært beslektede arter etter divergensen av hvete fra rug.
6. Undersøkelse av transkripsjonsfaktor (TF) og sykdomsresistensgener
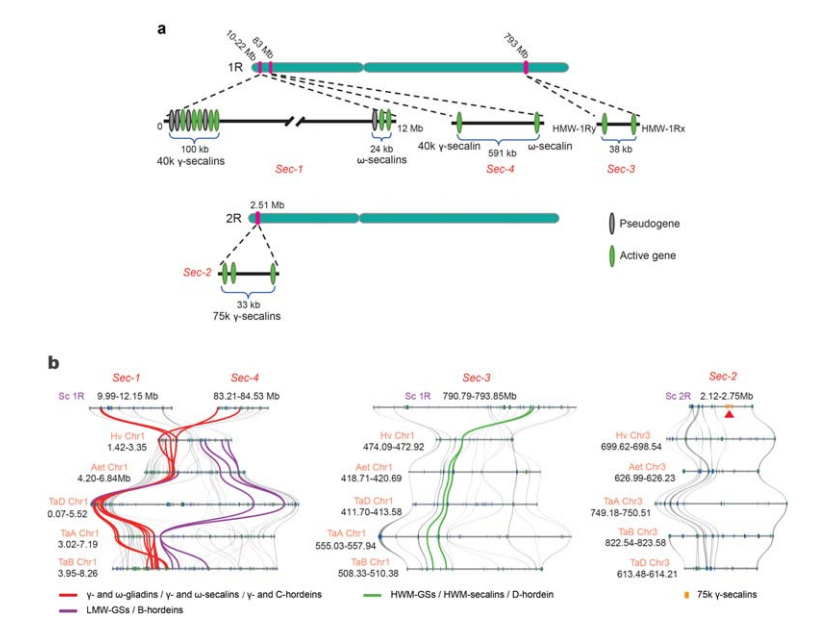
Analyse av rug secalin loci
Weining rug hadde flere sykdomsresistensassosierte (DRA) gener (1 989, tilleggsdata 3) enn Tu (1 621), Aet (1 758), Hv (1 508), Bd (1 178), Os (1 575) og A (1 836). ), B (1 728) og D (1 888) subgenomer av vanlig hvete.
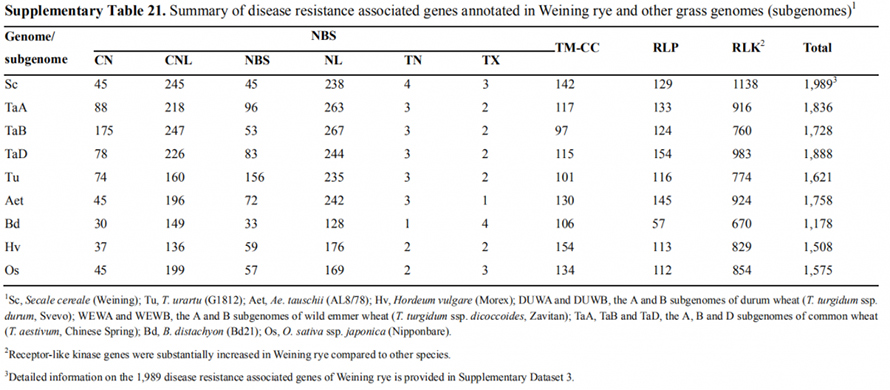
7. Undersøkelse av genekspresjonstrekk assosiert med tidlig overskriftstrekk
To FT-gener med relativt høy ekspresjon under langdagsforhold, ScFT1 og ScFT2, ble kommentert i Weining-genomsamlingen.To aminosyrerester av ScFT2 (S76 og T132) fosforylering ble funnet sammenheng med senkende tidskontroll
Utviklings- og genuttrykkstrekk assosiert med den tidlige overskriften til Weining rug
8. Gruvedrift av kromosomale områder og loki som potensielt er involvert i rugdomestisering
Totalt 123 647 SNP-er ble brukt til å utføre selevtive sweep-analyser mellom kultivert rug og S. vavilovii.11 selektive sveipesignaler identifisert ved reduksjonsindeks (DRI), fikseringsindeks (FST) og XP-CLR-metode.ScID1 ble funnet mulig involvering i reguleringen av overskriftsdato.
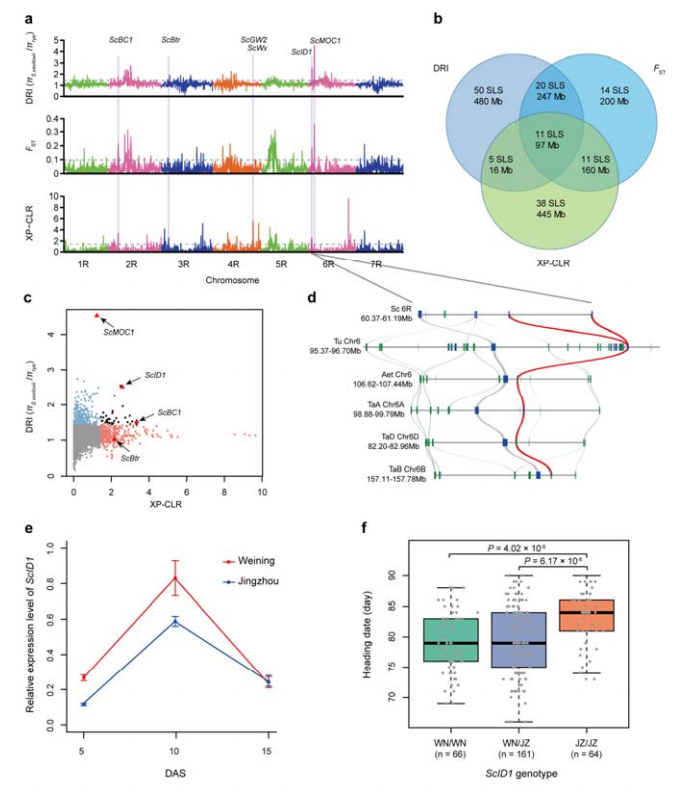
Identifikasjon og analyse av kromosomale regioner og loki potensielt relatert til rugdomestisering
Henvisning
Li GW et al.En genomsamling av høy kvalitet fremhever ruggenomiske egenskaper og agronomisk viktige gener.Naturgenetikk (2021)
Nyheter og høydepunkter tar sikte på å dele de siste vellykkede tilfellene med Biomarker Technologies, fange nye vitenskapelige prestasjoner samt fremtredende teknikker brukt under studien.
Innleggstid: Jan-05-2022