

EWOLUCJA GENOMEMU
genetyka natury
Wysokiej jakości zespół genomu podkreśla cechy genomiczne żyta i geny ważne z agronomicznego punktu widzenia
PacBio |Ilumina |Mapa optyczna Bionano |Zespół genomu Hi-C |Mapa Genetyczna |Selektywne przemiatanie |Sekwencja RNA |ISO-seq |SLAF-sekw
W ramach tego badania firma Biomarker Technologies zapewniła wsparcie techniczne w zakresie sekwencjonowania Pacbio, sekwencjonowania Hi-C i analizy danych.
Przegląd najważniejszych wydarzeń
1. Uzyskano pierwszy genom żyta wysokiej jakości na poziomie chromosomów, w którym pojedynczy chromosom ma rozmiar większy niż 1 Gb.
2. W porównaniu z genomami Tu, Aet i Hv, w genomie żyta zaobserwowano ostatnio wyjątkowe zdarzenia LTR-RT, które były odpowiedzialne za wydłużenie rozmiaru genomu żyta.
3. Rozbieżność między żytem a pszenicą diploidalną nastąpiła po oddzieleniu jęczmienia od pszenicy, przy czym czasy rozbieżności dla obu zdarzeń wynosiły w przybliżeniu 9,6 i 15 MYA.
Fosforylacja genów FT może kontrolować wczesną cechę kłoszenia żyta.
4. Analiza selektywnego przemiatania wskazuje na możliwy udział ScID1 w regulacji terminu kłosowania i prawdopodobną jego selekcję poprzez udomowienie u żyta
Tło
Tło
Żyto jest cenną rośliną spożywczą i pastewną, ważnym zasobem genetycznym do doskonalenia pszenicy i pszenżyta oraz niezbędnym materiałem do skutecznych badań porównawczych genomiki traw.Żyto weining, wczesna odmiana kwitnąca uprawiana w Chinach, wyróżnia się szerokim spektrum odporności zarówno na mączniaka prawdziwego, jak i rdzę pręgowaną.Aby zrozumieć genetyczne i molekularne podstawy elitarnych cech żyta oraz promować badania genomiczne i hodowlane żyta i roślin pokrewnych, zsekwencjonowaliśmy i przeanalizowaliśmy genom żyta Weining.
Osiągnięcia
Genom żyta
Genom żyta skonstruowano poprzez połączenie odczytów PacBio SMRT, sekwencjonowania krótkiego odczytu Illumina, a także odczytów z wychwytu konformacji chromatyny (Hi-C), mapowania genetycznego i analizy BioNano.Złożone kontigi (7,74 Gb) stanowiły 98,47% szacowanej wielkości genomu (7,86 Gb), przy czym 93,67% kontigów (7,25 Gb) przypisanych było do siedmiu chromosomów.Powtarzające się elementy stanowiły 90,31% złożonego genomu.
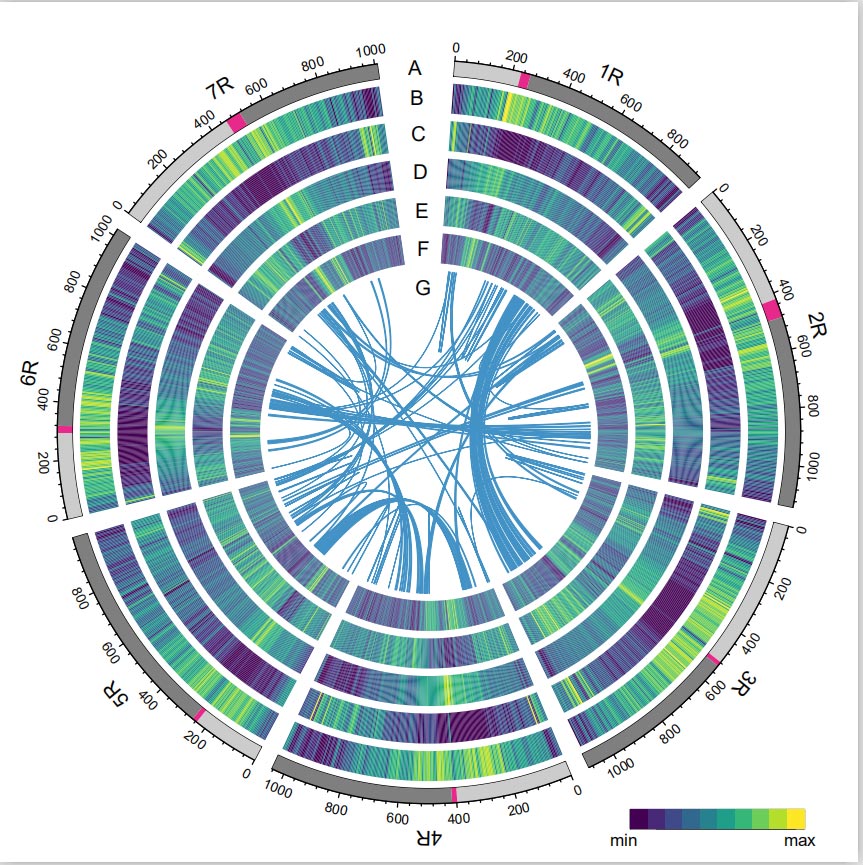
Genom żyta
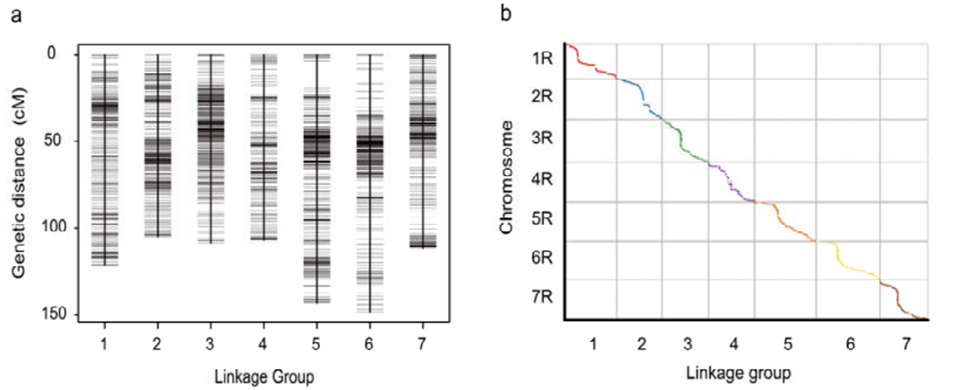
Mapa powiązań genetycznych (WJ) opracowana przy użyciu 295 roślin F2 pochodzących ze skrzyżowania dwóch odmian żyta zwyczajnego (Weining × Jingzhou)
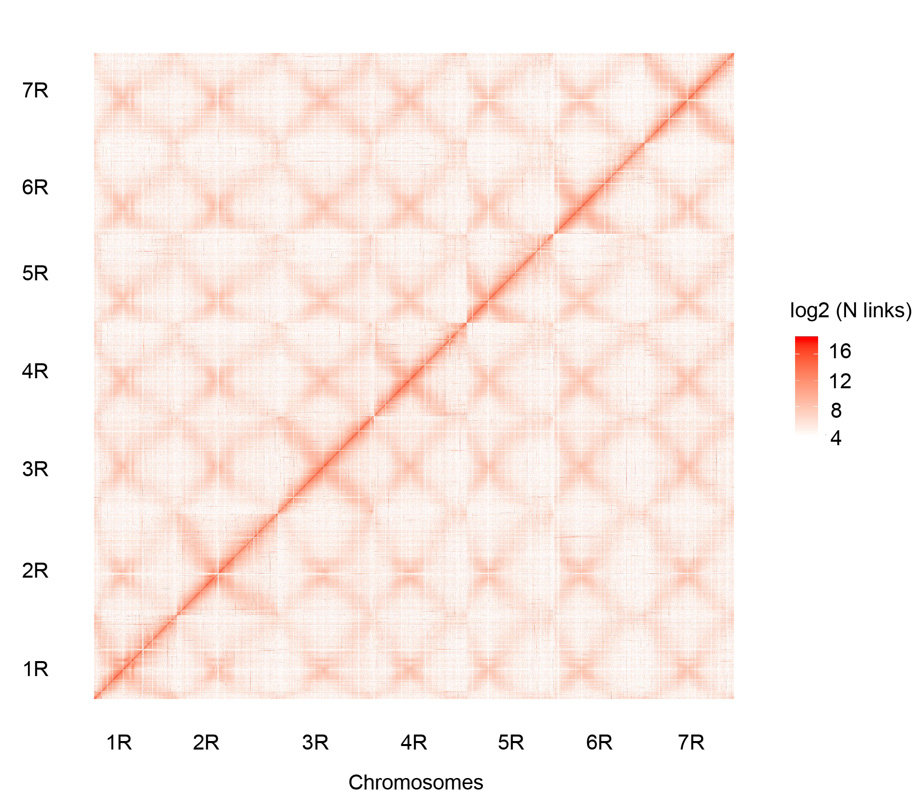
Mapa kontaktowa Hi-C siedmiu złożonych chromosomów żyta Weining (1R – 7R)
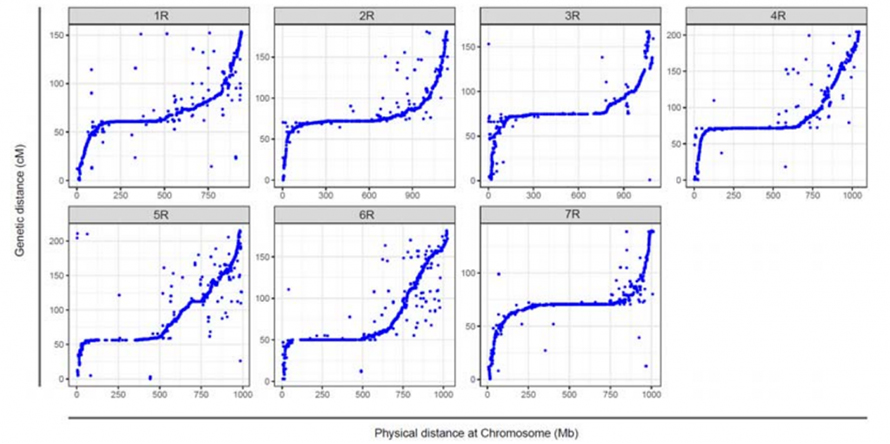
Dopasowanie pomiędzy siedmioma złożonymi chromosomami żyta Weining i siedmioma grupami połączeń żyta opracowanymi przy użyciu populacji Lo7 x Lo255 RIL
Stwierdzono, że wartość wskaźnika składania LTR (LAI) genomu żyta wynosi 18,42 i zidentyfikowano 1393 (96,74%) z 1440 wysoce konserwatywnych genów BUSCO. Wyniki te sugerują, że sekwencja genomu żyta Weininga jest wysokiej jakości zarówno w genach międzygenowych i regiony genowe.Przewidywano łącznie 86 991 genów kodujących białka, w tym 45 596 genów o wysokim stopniu pewności (HC) i 41 395 genów o niskim poziomie zaufania (LC).
2. Analiza TE
Analiza TE.Łącznie 6,99 Gb, co stanowi 90,31% zespołu Weininga, oznaczono jako TE, które obejmowały 2 671 941 elementów należących do 537 rodzin.Ta zawartość TE była wyraźnie wyższa niż wcześniej zgłaszana dla Ta (84,70%), Tu (81,42%), Aet (84,40%), WEW (82,20%) lub Hv (80,80%).Retrotranspozony z długimi końcowymi powtórzeniami (LTR-RT), w tym elementy Gypsy, Copia i niesklasyfikowane RT, były dominującymi TE i zajmowały 84,49% opisanej zawartości TE i 76,29% złożonego genomu Weininga;Transpozony DNA CACTA były drugimi pod względem liczebności TE, stanowiąc 11,68% opisanej zawartości TE i 10,55% złożonego genomu Weininga.
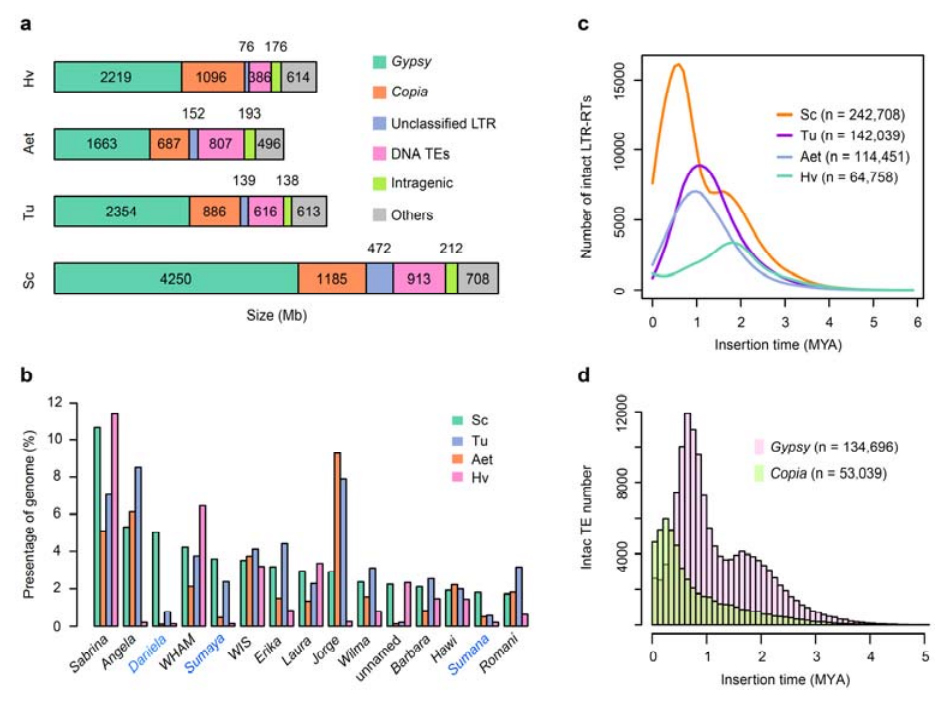
Analiza elementów transpozonowych żyta
W żyto odrobaczanym występował stosunkowo wysoki odsetek niedawnych insercji LTR-RT, przy czym szczyt amplifikacji pojawił się około 0,5 miliona lat temu (MYA), co było najpóźniejszym spośród czterech gatunków;drugi pik, wystąpił około 1,7 MYA, był starszy i również występował w jęczmieniu.Na poziomie nadrodziny znaleziono bardzo niedawne wybuchy pierwiastków Copia w żyto Weining przy 0,3 MYA, podczas gdy amplifikacje cygańskich RT w dominujący sposób ukształtowały bimodalny wzór dystrybucji dynamiki wybuchu LTR-RT.
3. Badanie ewolucji genomu żyta i syntonii chromosomów
Rozbieżność między żytem a pszenicą diploidalną nastąpiła po oddzieleniu jęczmienia od pszenicy, przy czym czasy rozbieżności dla obu zdarzeń wynosiły odpowiednio około 9,6 i 15 MYA.1R, 2R, 3R były całkowicie współliniowe odpowiednio z chromosomami grup 1, 2 i 3 pszenicy.Stwierdzono, że 4R, 5R, 6R, 7R istnieją fuzje i segmenty na dużą skalę.
4. Analiza duplikacji genów i ich wpływu na geny biosyntezy skrobi
Warto zauważyć, że liczba genów zduplikowanych tandemowo (TDG) i genów zduplikowanych proksymalnie (PDG) żyta Weining była wyższa niż liczba znaleziona dla Tu, Aet, Hv, Bd i Os.Transponowane zduplikowane geny (TrDG) były również liczniejsze niż te specyficzne dla Tu i Aet.Ekspansji genomu żyta towarzyszy większa liczba duplikacji genów.Zwiększone wybuchy TE w życie mogły prowadzić do podwyższonej liczby TrDG.
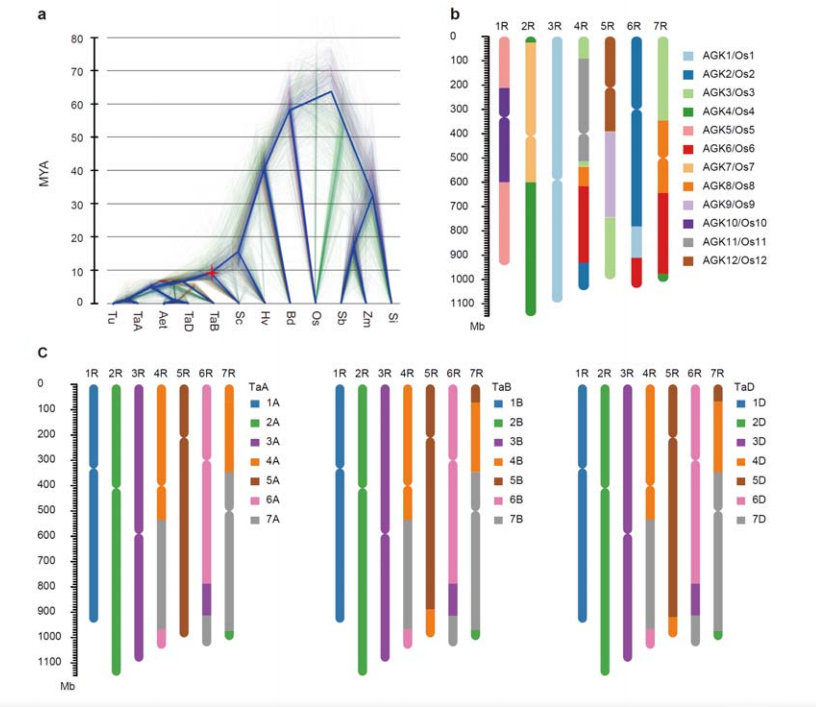
Analizy ewolucyjne i synteny chromosomów genomu żyta
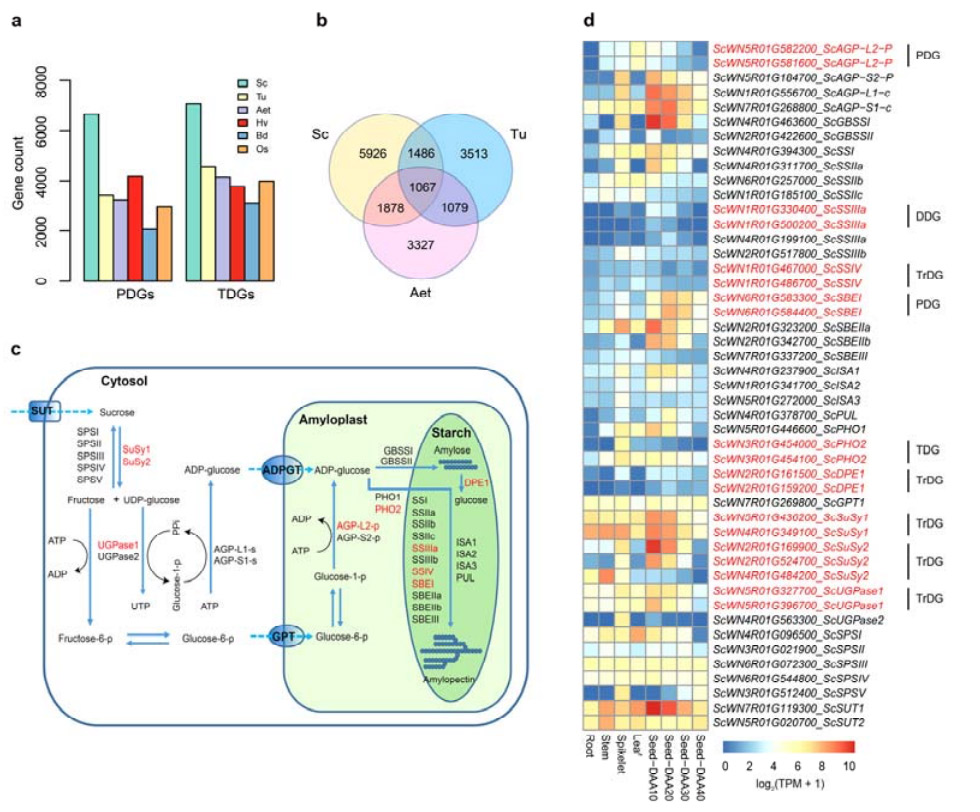
Analiza duplikacji genów żyta i ich wpływ na różnorodność genów związanych z biosyntezą skrobi (SBRG)
5. Sekcja loci genów białka spichrzowego nasion żyta (SSP).
Na 1R lub 2R zidentyfikowano cztery loci chromosomalne (Sec-1 do Sec-4) określające SSP żyta.Geny α-gliadyny wyewoluowały dopiero niedawno u pszenicy i gatunków blisko spokrewnionych, po oddzieleniu się pszenicy od żyta.
6. Badanie czynników transkrypcyjnych (TF) i genów odporności na choroby
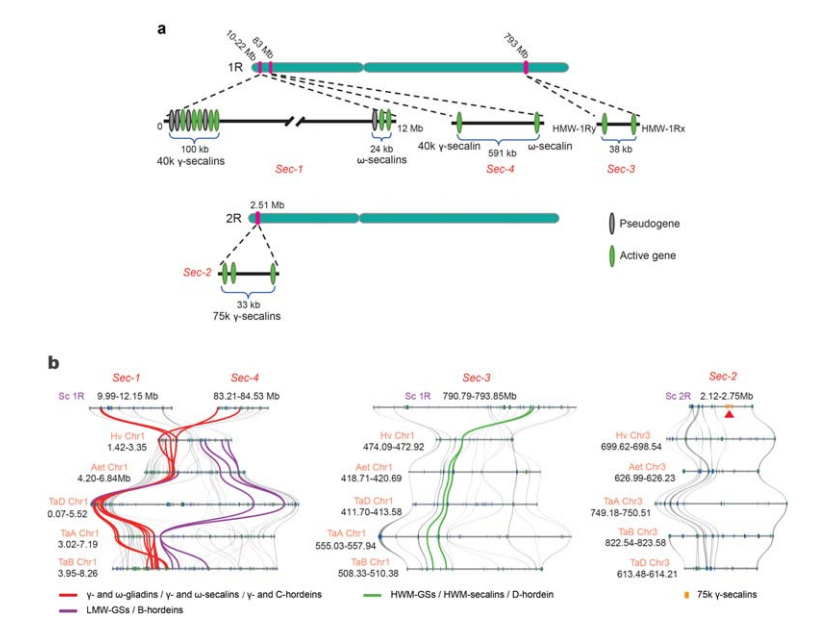
Analiza loci sekaliny żyta
Żyto odsadzające miało liczniejsze geny związane z odpornością na choroby (DRA) (1989, dane uzupełniające 3) niż Tu (1621), Aet (1758), Hv (1508), Bd (1178), Os (1575) i A (1836) ), B (1728) i D (1888) subgenomów pszenicy zwyczajnej.
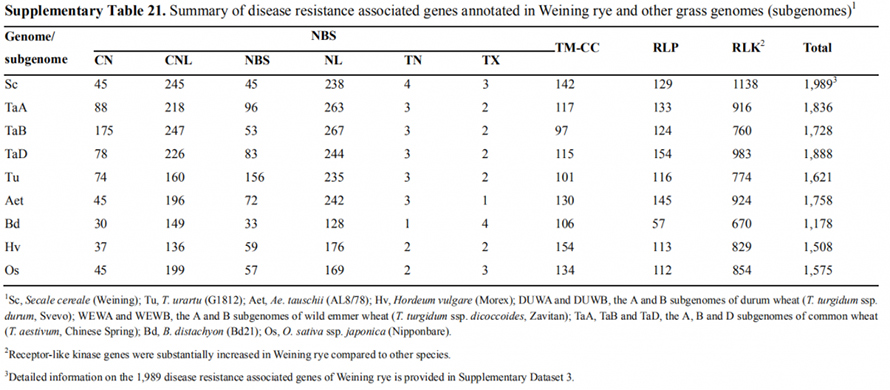
7. Badanie cech ekspresji genów związanych z wczesną cechą główkowania
Podczas składania genomu Weininga odnotowano dwa geny FT o stosunkowo wysokiej ekspresji w warunkach długiego dnia, ScFT1 i ScFT2.Stwierdzono związek dwóch reszt aminokwasowych fosforylacji ScFT2 (S76 i T132) z kontrolą czasu obniżania
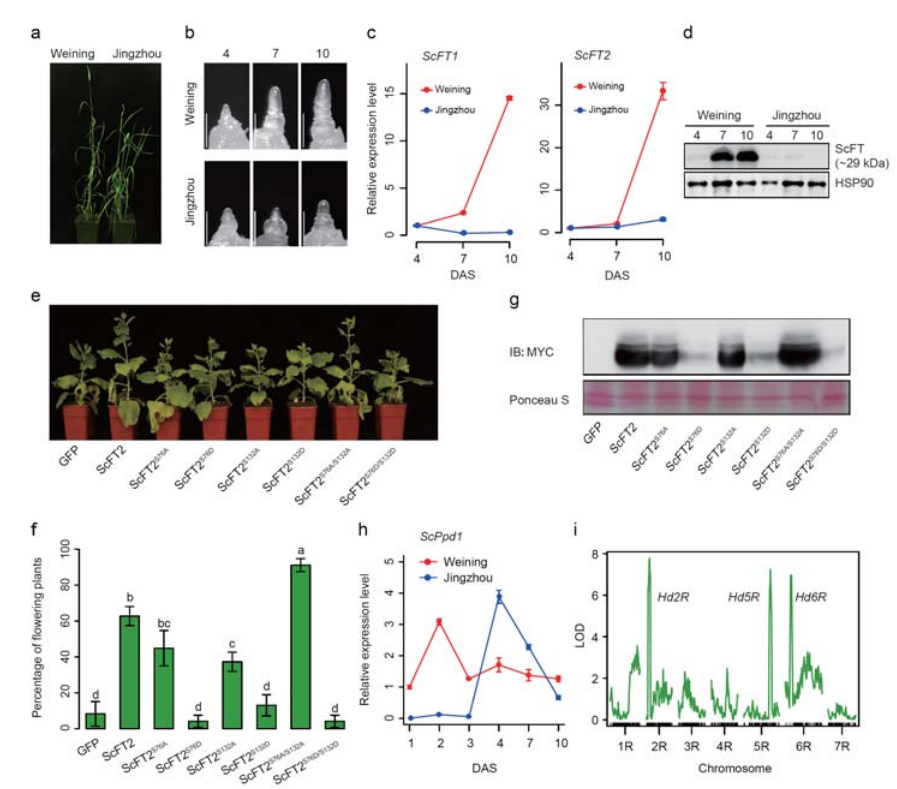
Cechy rozwojowe i ekspresja genów związane z cechą wczesnego kłosowania żyta Weining
8. Eksploatacja regionów i loci chromosomalnych potencjalnie zaangażowanych w udomowienie żyta
Do przeprowadzenia selektywnej analizy zamiatania żyta uprawnego i S. vavilovii wykorzystano łącznie 123 647 SNP.11 selektywnych sygnałów przemiatania identyfikowanych za pomocą wskaźnika redukcji (DRI), wskaźnika fiksacji (FST) i metody XP-CLR.Stwierdzono, że ScID1 może mieć wpływ na regulację daty kursu.
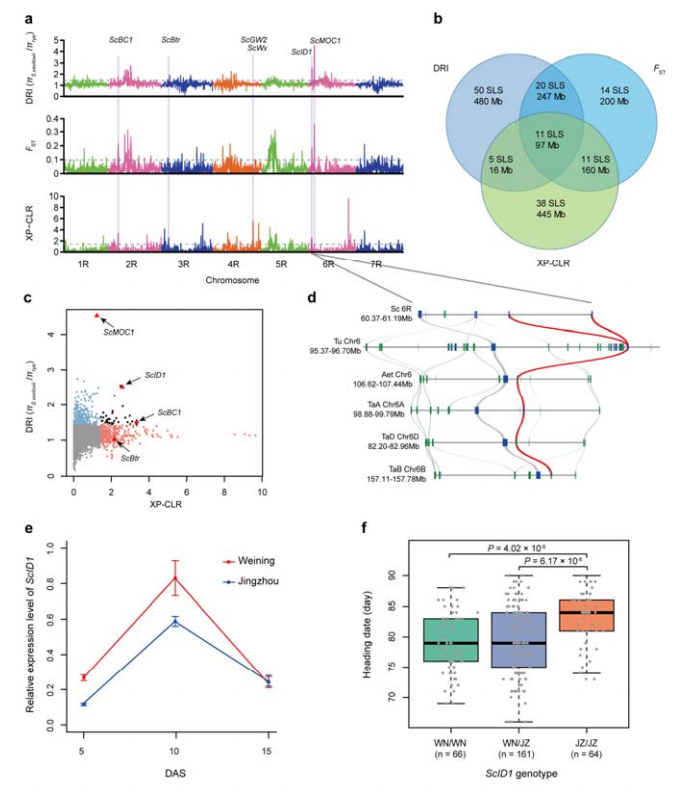
Identyfikacja i analiza regionów i loci chromosomalnych potencjalnie związanych z udomowieniem żyta
Odniesienie
Li GW i in.Wysokiej jakości zespół genomu podkreśla cechy genomiczne żyta i geny ważne z agronomicznego punktu widzenia.Genetyka natury (2021)
Wiadomości i najważniejsze informacje ma na celu podzielenie się najnowszymi pomyślnymi przypadkami z firmą Biomarker Technologies, uchwycenie nowatorskich osiągnięć naukowych, a także wybitnych technik zastosowanych podczas badania.
Czas publikacji: 05 stycznia 2022 r