

Zbiorcza analiza Segreganta






Zalety serwisu
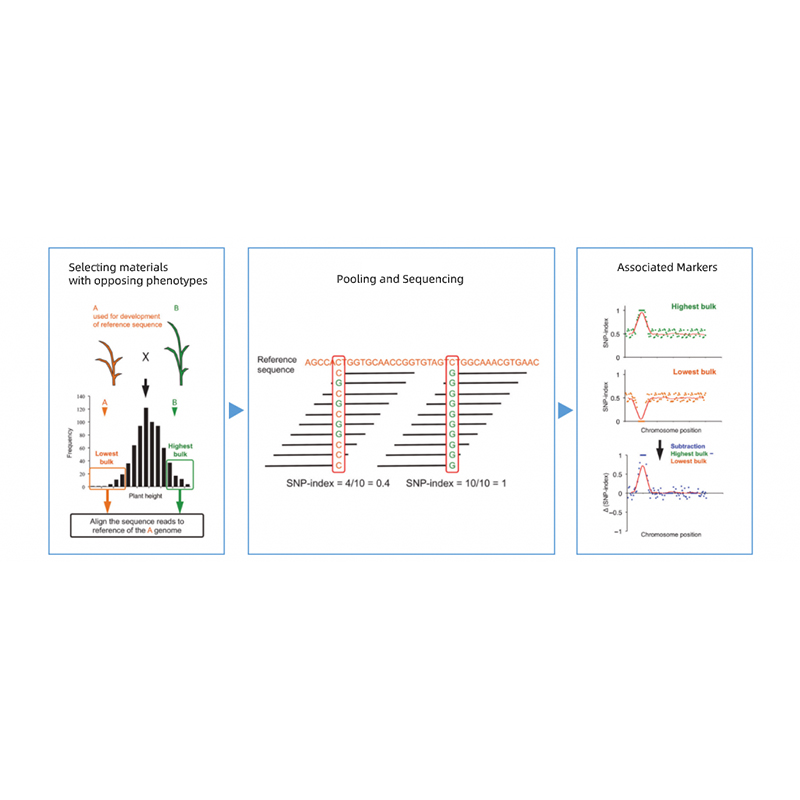
Takagi i wsp., The Plant Journal, 2013
● Dokładna lokalizacja: Mieszanie grup od 30+30 do 200+200 osobników w celu zminimalizowania hałasu w tle;przewidywanie regionu kandydującego na podstawie niesynonimicznych mutantów.
● Kompleksowa analiza: szczegółowa adnotacja dotycząca funkcji genu kandydującego, w tym NR, SwissProt, GO, KEGG, COG, KOG itp.
● Szybszy czas realizacji: Szybka lokalizacja genu w ciągu 45 dni roboczych.
● Rozległe doświadczenie: BMK przyczyniło się do lokalizacji tysięcy cech, obejmujących różnorodne gatunki, takie jak rośliny uprawne, produkty wodne, lasy, kwiaty, owoce itp.
Specyfikacje usług
Populacja:
Segregacja potomstwa rodziców o przeciwstawnych fenotypach.
np. potomstwo F2, krzyżowanie wsteczne (BC), rekombinowana linia wsobna (RIL)
Basen do mieszania
Dla cech jakościowych: 30 do 50 osobników (minimum 20)/masę
W przypadku tratis ilościowych: górne 5% do 10% osobników z którymkolwiek skrajnym fenotypem w całej populacji (minimum 30+30).
Zalecana głębokość sekwencjonowania
Co najmniej 20X/rodzic i 1X/osobnik potomstwa (np. w przypadku puli mieszanej potomstwa składającej się z 30+30 osobników, głębokość sekwencjonowania będzie wynosić 30X na partię)
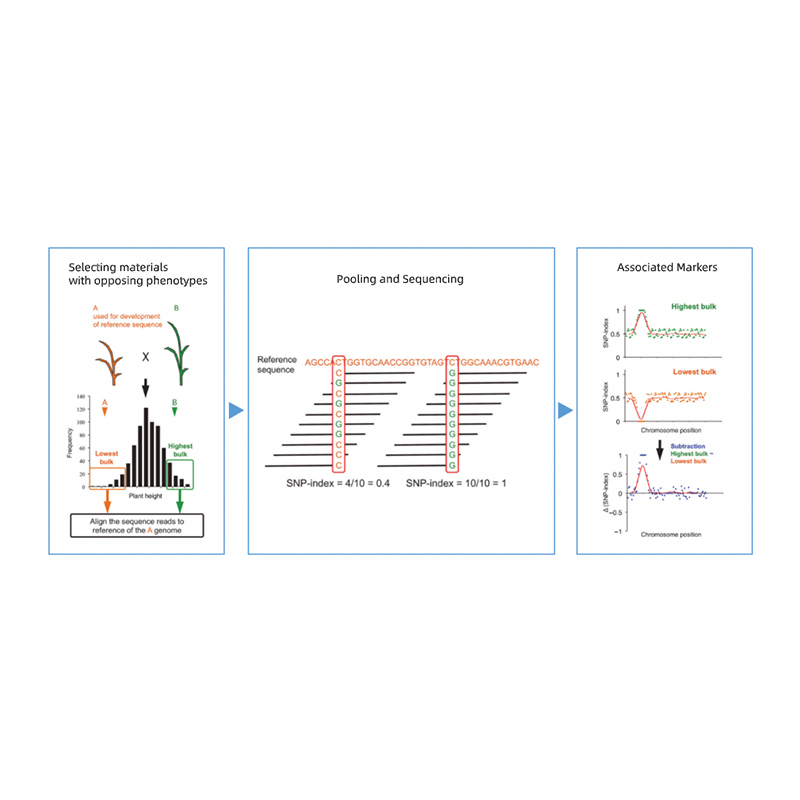
Analizy bioinformatyczne
● Ponowne sekwencjonowanie całego genomu
● Przetwarzanie danych
● Połączenia SNP/Indel
● Kontrola regionu kandydującego
● Adnotacja dotycząca funkcji genu kandydata
Przykładowe wymagania i dostawa
Przykładowe wymagania:
Nukleotydy:
| próbka gDNA | Próbka tkanki |
| Stężenie: ≥30 ng/μl | Rośliny: 1-2 g |
| Ilość: ≥2 μg (objętość ≥15 μl) | Zwierzęta: 0,5-1 g |
| Czystość: OD260/280= 1,6-2,5 | Krew pełna: 1,5 ml |
Przebieg prac serwisowych

Projekt eksperymentu

Dostawa próbek

Ekstrakcja RNA

Budowa biblioteki

Sekwencjonowanie

Analiza danych

Usługi posprzedażowe
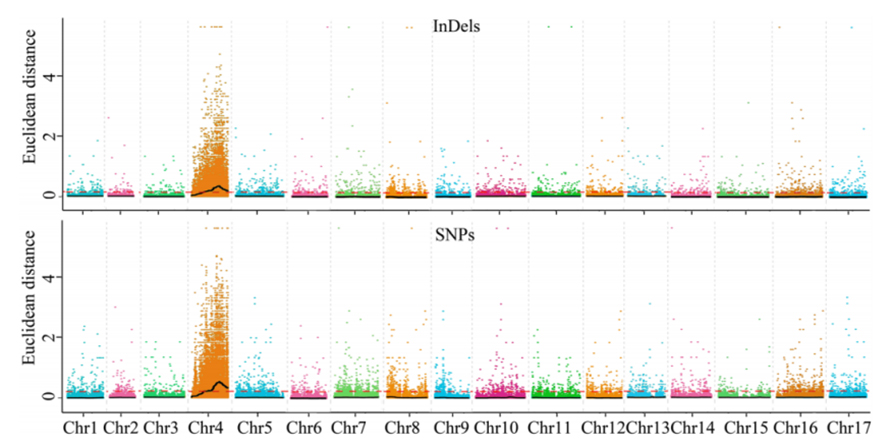
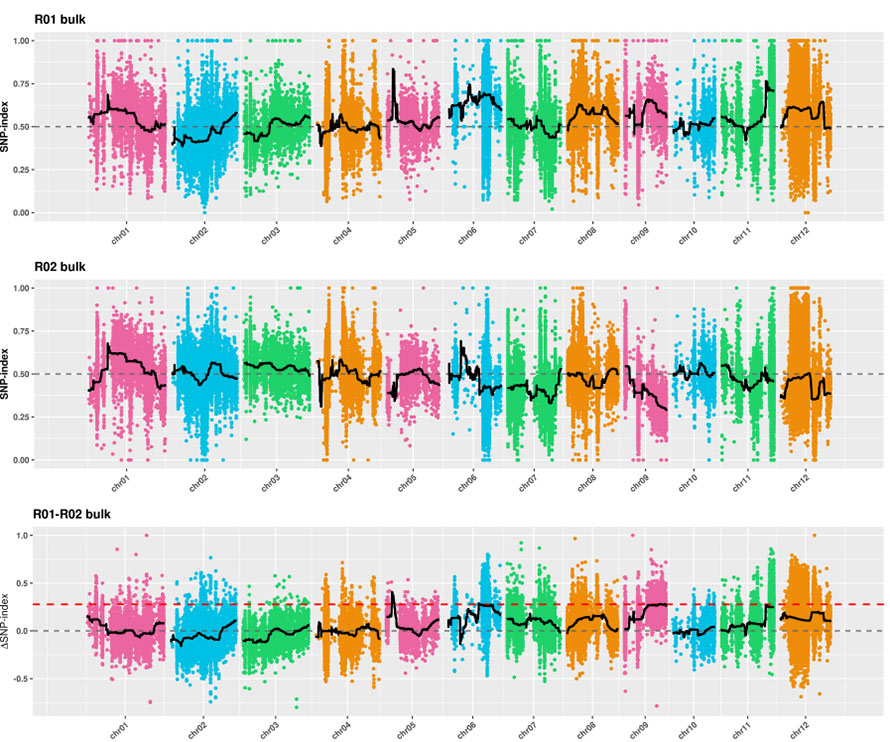
1. Analiza skojarzeń oparta na odległości euklidesowej (ED) w celu zidentyfikowania regionu kandydującego.Na poniższym rysunku
Oś X: liczba chromosomów;Każda kropka reprezentuje wartość ED SNP.Czarna linia odpowiada dopasowanej wartości ED.Wyższa wartość ED wskazuje na bardziej znaczący związek między miejscem a fenotypem.Czerwona linia przerywana przedstawia próg znaczącego powiązania.
2.Analiza skojarzeń nie oparta na indeksie SNP
Oś X: liczba chromosomów;Każda kropka reprezentuje wartość indeksu SNP.Czarna linia oznacza dopasowaną wartość indeksu SNP.Im większa wartość, tym bardziej znaczące jest powiązanie.
Sprawa BMK
Locus cechy ilościowej Fnl7.1 o głównym efekcie koduje obfite białko późnej embriogenezy związane z długością szyjki owocowej u ogórka
Opublikowany: Dziennik biotechnologii roślin, 2020
Strategia sekwencjonowania:
Rodzice (Jin5-508, YN): Ponowne sekwencjonowanie całego genomu 34× i 20×.
Pule DNA (50 osób z długą szyją i 50 z krótką szyją): Ponowne sekwencjonowanie dla 61× i 52×
Kluczowe wyniki
W tym badaniu populację segregującą (F2 i F2:3) wygenerowano poprzez skrzyżowanie linii ogórka długoszyjnego Jin5-508 i YN o krótkiej szyi.Dwie pule DNA zostały skonstruowane przez 50 osobników o wyjątkowo długiej szyi i 50 osobników o skrajnie krótkiej szyi.QTL z głównym efektem zidentyfikowano na Chr07 za pomocą analizy BSA i tradycyjnego mapowania QTL.Region kandydujący został dodatkowo zawężony poprzez dokładne mapowanie, ilościowe oznaczanie ekspresji genów i eksperymenty na transgenikach, które ujawniły gen kluczowy do kontrolowania długości szyi, CsFnl7.1.Ponadto stwierdzono, że polimorfizm w regionie promotora CsFn17.1 jest powiązany z odpowiednią ekspresją.Dalsza analiza filogenetyczna sugeruje, że locus Fnl7.1 najprawdopodobniej pochodzi z Indii.
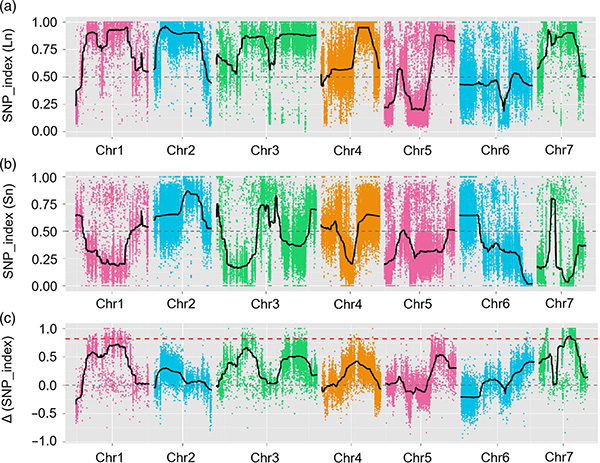 Mapowanie QTL w analizie BSA w celu zidentyfikowania regionu kandydującego powiązanego z długością szyjki ogórka | 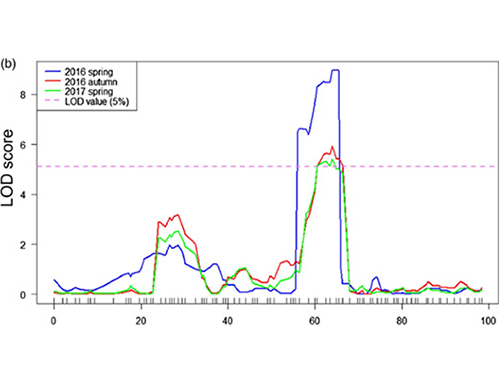 Profile LOD QTL o długości szyi ogórka zidentyfikowane na Chr07 |
Xu, X. i in.„Locus cechy ilościowej Fnl7.1 o głównym efekcie koduje obfite białko późnej embriogenezy, powiązane z długością szyjki owocowej u ogórka”.Dziennik Biotechnologii Roślin 18.7 (2020).





