

GENOMIA CZŁOWIEKA
genetyka natury
Sekwencjonowanie z długim odczytem identyfikuje ekspansje powtórzeń GGC w NOTCH2NLC związane z chorobą wtrętów wewnątrzjądrowych neuronów
Ponowne sekwencjonowanie ONT |Ilumina |Sekwencjonowanie całego egzomu |Ukierunkowane sekwencjonowanie CRISPR-Cas9 ONT |Sekwencja RNA |Wywołanie metylacji ONT 5mC
Przegląd najważniejszych wydarzeń
1. Dzięki analizie powiązań dużej rodziny NIID zidentyfikowano dwa powiązane regiony.
2. Sekwencjonowanie z długim odczytem w oparciu o ONT i wzbogacanie za pośrednictwem Cas-9. Sekwencjonowanie ONT odkryło potencjalną genetyczną przyczynę ekspansji powtórzeń NIID i GGC w 5′ UTR NOTCH2NLC.W badaniu tym po raz pierwszy odnotowano powtarzające się ekspansje genów specyficznych dla człowieka, które ewoluowały poprzez duplikacje segmentowe.
3. Sekwencjonowanie RNA ujawniło nieprawidłowe transkrypty antysensowne na początku lub wewnątrz regionów ekspansji powtórzeń GGC w NOTCH2NLC.
Tło
Nchoroba wtrętów wewnątrzjądrowych euronału (NIID) jest postępującą i śmiertelną chorobą neurodegeneracyjną, która charakteryzuje się obecnością eozynofilowych, szklistych wtrętów wewnątrzjądrowych w ośrodkowym i obwodowym układzie nerwowym.Jej bardzo zmienna manifestacja kliniczna stwarza duże trudności diagnostyczne do czasu wprowadzenia biopsji skóry.Jednakże metody oparte na histopatologii nadal spotykają się z błędną diagnozą, co wymaga genetycznego zrozumienia NIID.
Osiągnięcia
Analiza powiązań
SSekwencjonowanie całego genomu (WGS) i sekwencjonowanie całego egzomu (WES) przeprowadzono na dużej rodzinie NIID (13 członków dotkniętych chorobą i 7 zdrowych członków).Analiza powiązań SNP wyodrębnionych z tych danych ujawniła tylko dwa połączone regiony: region 3,5 Mb przy 1p36.31-p36.22 (maksymalna LOD = 2,32) i region 58,1 Mb przy 1p22.1-q21.3 (maksymalna LOD: 4,21 ).Jednakże w tych połączonych regionach nie zidentyfikowano żadnych patogennych SNP ani CNV.
Powtarzające się rozszerzenia GGC w NOTCH2NLC
NSekwencjonowanie w oparciu o anopore przeprowadzono u 13 chorych i 4 zdrowych członków z 8 rodzin (inny członek dotknięty chorobą został zsekwencjonowany za pomocą platformy sekwencjonowania o długim odczytaniu Pacbio).Długoterminowe dane ujawniły związaną z chorobą ekspansję powtórzeń GGC w 5′ UTR mapowania genu NOTCH2NLC na region połączony o wielkości 58,1 Mb (Rysunek 1).Te powtarzające się ekspansje zidentyfikowano także we wszystkich 40 sporadycznych przypadkach NIID testowanych metodą RP-PCR.
CZastosowano sekwencjonowanie docelowe za pośrednictwem as-9 na platformie nanoporów, aby uzyskać większe pokrycie odczytu powtórzeń NOTCH2NLC (100 X-1795 X).Te sekwencje konsensusowe dobrze zgadzały się z wcześniejszymi ustaleniami dotyczącymi rozszerzeń powtórzeń GGC.Co więcej, zidentyfikowano powtórzenia {(GGA)n (GGC)n}n jako potencjalny marker genetyczny fenotypu z dominacją osłabienia (ryc. 2).
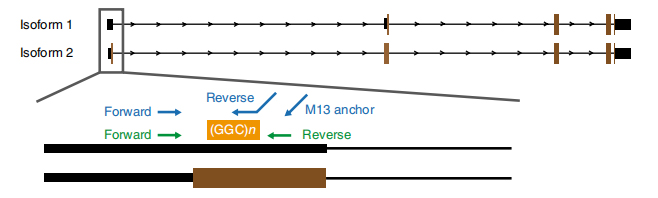
Rycina 1. Ekspansja powtórzeń związana z chorobą zidentyfikowana w eksonie 1 izoform NOTCH2NLC.
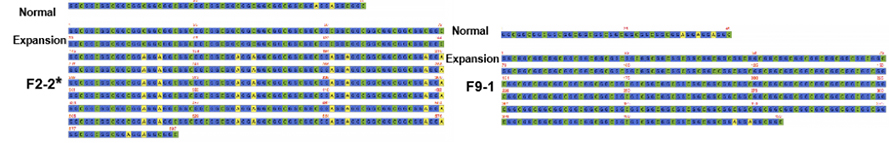
Rycina 2. Zgodne sekwencje powtórzeń NPTCH2NLC u pacjentów z NIID z (*) lub bez fenotypu dominującego osłabienia
NGeny OTCH2NL to geny specyficzne dla człowieka, które, jak się uważa, odgrywają kluczową rolę w ewolucji ludzkiego mózgu i chorobach neurologicznych.Jednakże trzy geny powiązane z NOTCH2 (NOTCH2NLA, NOTCH2NLB i NOTCH2NLC) o > 99,1% identyczności sekwencji nie zostały rozwiązane aż do najnowszego złożenia ludzkiego genomu.Wolne od syntezy sekwencjonowanie z długim odczytem na platformie nanoporów wykazało zauważalne korzyści w rozdzielaniu regionów o wysokim podobieństwie i powtórzeniach (GGC)n ze 100% bogactwem GC.
Powtarzające się rozszerzenia GGC w NOTCH2NLC
TSekwencjonowanie ranscriptomu przeprowadzono na 2 dotkniętych i 2 zdrowych członkach.Znormalizowaną głębokość odczytu obliczono dla nici sensownej i antysensownej w górę od pierwszych eksonów paralogów NOTCH2NL.Nieprawidłowe transkrypty antysensowne znaleziono tylko w dotkniętych przypadkach, które znajdują się na początku lub w obszarze powtarzalnej ekspansji (fioletowe piki w F1-14 i F1-16 na Rycinie 3.).Ponadto zidentyfikowano 54 DEG i wszystkie zostały wzbogacone o terminy GO i MPO związane z funkcjami neuronowymi.
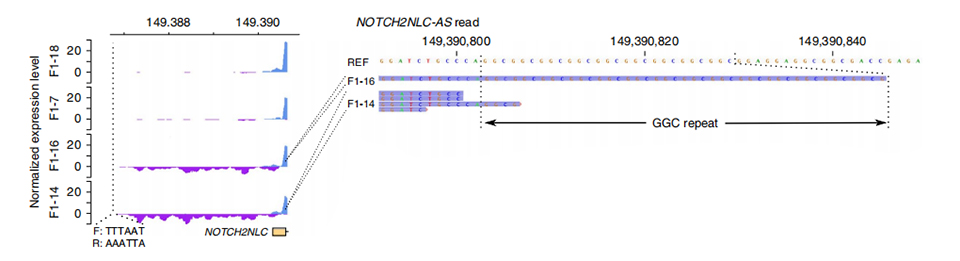
Rycina 3. Znormalizowana głębokość odczytu powyżej pierwszego eksonu NOTCH2NLC w przypadkach nienaruszonych (powyżej) i dotkniętych (poniżej).
Technologia
Technologia Oxford Nanopore (ONT)
NSekwencjonowanie anopore różni się od innych platform sekwencjonowania tym, że nukleotydy są odczytywane bezpośrednio, bez procesu syntezy DNA.Gdy pojedyncza nić DNA przechodzi przez pory białkowe (nanopory) wielkości nanometrów, różne nukleotydy generują różne prądy jonowe, które można wychwycić i przenieść do sekwencji zasad.Sama platforma sekwencjonowania ONT nie wykazuje widocznych ograniczeń technicznych dotyczących długości odczytu DNA.Dlatego dostępne są ultradługie odczyty (ULR), umożliwiające składanie genomu o wysokiej jakości.Co więcej, te niezwykle długie odczyty, które są wystarczająco długie, aby przeciąć złożone cechy sekwencji lub zmiany strukturalne, pomagają przezwyciężyć tutaj ograniczenia sekwencjonowania z krótkim odczytem.

Sekwencjonowanie nanoporów
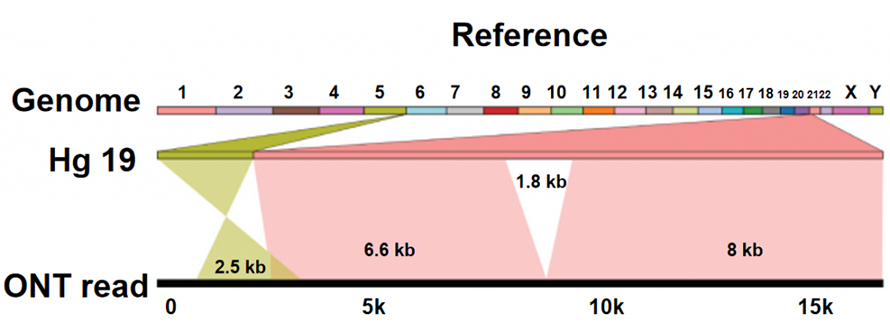
Identyfikacja zmienności struktury (SV).
SSekwencjonowanie wolne od syntezy w dużym stopniu zachowuje informację o metylacji DNA na szablonie.Metylowane A, T, C i G generują odrębne prądy jonowe od niemetylowanych, które można odczytać bezpośrednio przez platformę.Sekwencjonowanie nanoporów umożliwia profilowanie całego genomu zarówno przy 5 mC, jak i 6 mA przy rozdzielczości pojedynczego nukleotydu.
Odniesienie
Jun Sone i in.glin.Sekwencjonowanie z długim odczytem identyfikuje ekspansje powtórzeń GGC w NOTCH2NLC związane z neuronalną chorobą inkluzyjną wewnątrzjądrową.Genetyka natury (2019)
Technologia i najważniejsze informacje ma na celu podzielenie się najnowszymi pomyślnymi zastosowaniami różnych technologii sekwencjonowania o dużej przepustowości w różnych obszarach badawczych, a także genialnymi pomysłami w projektowaniu eksperymentów i eksploracji danych.
Czas publikacji: 06 stycznia 2022 r