
-

Hi-C based Chromatin Interaction
Hi-C is a method designed to capture genomic configuration by combining probing proximity-based interactions and high-throughput sequencing. The method is based on chromatin crosslinking with formaldehyde, followed by digestion and re-ligation in a way that only fragments that are covalently linked will form ligation products. By sequencing these ligation products, it is possible to study the 3D organization of the genome. Hi-C enables studying the distribution of the portions of the genome that are lightly packed (A compartments, euchromatin) and more likely to be transcriptionally active, and the regions that are more tightly packed (B compartments, Heterochromatin). Hi-C can also be used to pinpoint Topologically Associated Domains (TADs), regions of the genome that have folded structures and are likely to have similar expression patterns, and to identify chromatin loops, DNA regions that are anchored together by proteins and that are often enriched in regulatory elements. BMKGene’s Hi-C sequencing service empowers researchers to explore the spatial dimensions of genomics, opening new avenues for understanding genome regulation and its implications in health and disease.
-

Chromatin Immunoprecipitation Sequencing (ChIP-seq)
Chromatin Immunoprecipitation (CHIP) is a technique that leverages antibodies to selectively enrich DNA-binding proteins and their corresponding genomics targets. Its integration with NGS enables the genome-wide profiling of DNA targets associated with histone modification, transcription factors, and other DNA-binding proteins. This dynamic approach enables comparisons of binding sites across diverse cell types, tissues, or conditions. ChIP-Seq’s applications span from studying transcriptional regulation and developmental pathways to elucidating disease mechanisms, making it an indispensable tool for understanding genomic regulation landscapes and advancing therapeutic insights.
Platform: Illumina NovaSeq
-

Whole genome bisulfite sequencing(WGBS)
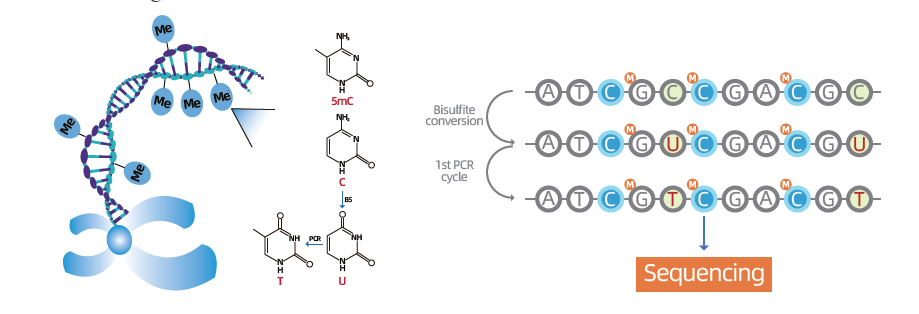
Whole Genome Bisulfite Sequencing (WGBS) stands as the gold-standard methodology for in-depth exploration of DNA methylation, specifically the fifth position in cytosine (5-mC), a pivotal regulator of gene expression and cellular activity. The principle underlying WGBS involves bisulfite treatment, inducing the conversion of unmethylated cytosines to uracil (C to U), while leaving methylated cytosines unchanged. This technique offers single-base resolution, allowing researchers to comprehensively investigate the methylome and uncover abnormal methylation patterns associated with various conditions, notably cancer. By employing WGBS, scientists can gain unparalleled insights into genome-wide methylation landscapes, providing a nuanced understanding of the epigenetic mechanisms that underlie diverse biological processes and diseases.
-

Assay for Transposase-Accessible Chromatin with High Throughput Sequencing (ATAC-seq)
ATAC-seq is a high-throughput sequencing technique used for genome-wide chromatin accessibility analysis. It use provides deeper understanding of the complex mechanisms of global epigenetic control over gene expression. The method uses a hyperactive Tn5 transposase to simultaneously fragment and tag open chromatin regions by inserting sequencing adaptors. Subsequent PCR amplification results in the creation of a sequencing library, which allows for the comprehensive identification of open chromatin regions under specific space-time conditions. ATAC-seq provides a holistic view of accessible chromatin landscapes, unlike methods that solely focus on transcription factor binding sites or specific histone-modified regions. By sequencing these open chromatin regions, ATAC-seq reveals regions more likely to active regulatory sequences and potential transcription factor binding sites, offering valuable insights into the dynamic modulation of gene expression across the genome.
-

Reduced Representation Bisulfite Sequencing (RRBS)

Reduced Representation Bisulfite Sequencing (RRBS) has emerged as a cost-effective and efficient alternative to Whole Genome Bisulfite Sequencing (WGBS) in DNA methylation research. While WGBS provides comprehensive insights by examining the entire genome at single base resolution, its high cost can be a limiting factor. RRBS strategically mitigates this challenge by selectively analyzing a representative portion of the genome. This methodology relies on the enrichment of CpG island-rich regions by MspI cleavage followed by size selection of 200-500/600 bps fragments. Consequently, only regions proximal to CpG islands are sequenced, while those with distant CpG islands are excluded from the analysis. This process, combined with bisulfite sequencing, allows for high-resolution detection of DNA methylation, and the sequencing approach, PE150, focuses specifically on the ends of the inserts rather than the middle, increasing the efficiency of methylation profiling. The RRBS is an invaluable tool that enables cost-effective DNA methylation research and advances knowledge of epigenetic mechanisms.
