Whole Transcriptome Sequencing – Illumina

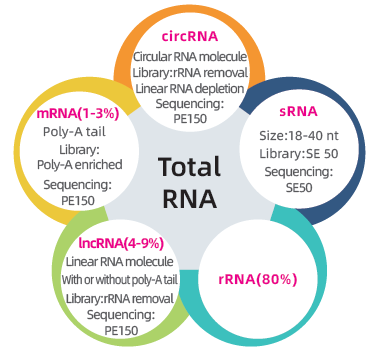
Features
● Dual library to sequence the complete transcriptome: rRNA depletion followed by PE150 library preparation and size selection followed by SE50 library preparation
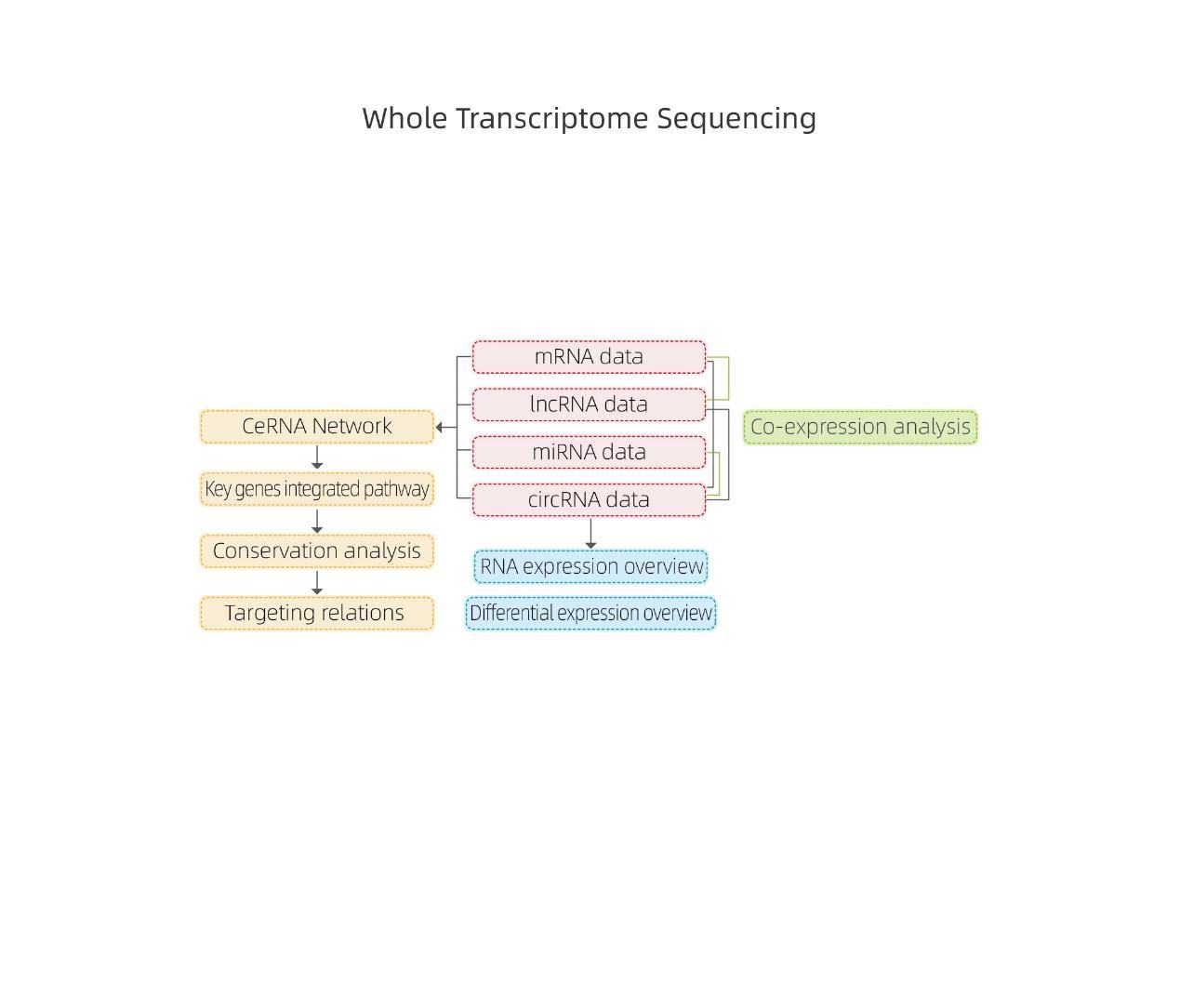
● Complete bioinformatics analysis of mRNA, lncRNA, circRNA, and miRNA in separate bioinformatics reports
● Joint analysis of all RNA expression in a combined report, including ceRNA networks analysis.
Service Advantages
● In-depth Analysis of Regulatory Networks: ceRNA network analysis is enabled by the joint sequencing of mRNA, lncRNA, circRNA, and miRNA and by an exhaustive bioinformatic workflow.
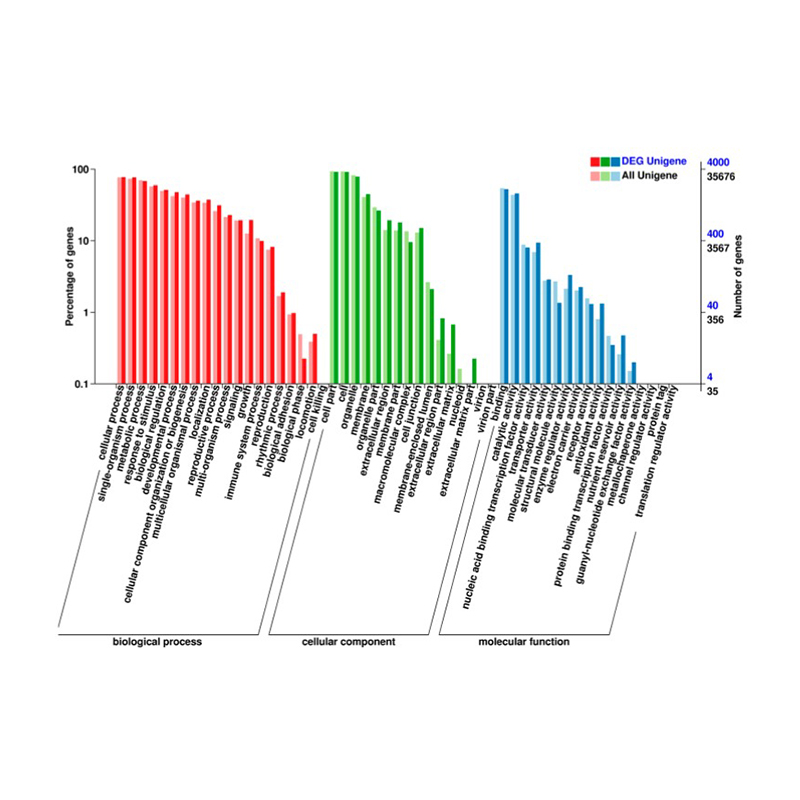
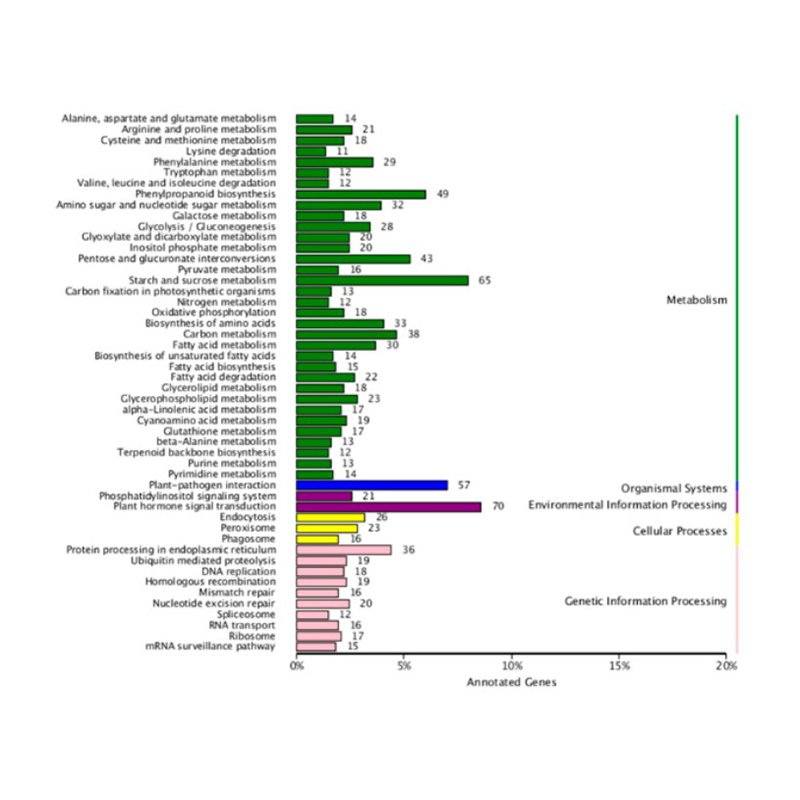
● Comprehensive Annotation: We use multiple databases to functionally annotate the Differentially Expressed Genes (DEGs) and perform the corresponding enrichment analysis, providing insights into the cellular and molecular processes underlying the transcriptome response.
● Extensive Expertise: With a track record of successfully closing over 2000 whole transcriptome projects in various research domain, our team brings a wealth of experience to every project.
● Rigorous Quality Control: We implement core control points across all stages, from sample and library preparation to sequencing and bioinformatics. This meticulous monitoring ensures the delivery of consistently high-quality results.
● Post-Sales Support: Our commitment extends beyond project completion with a 3-month after-sale service period. During this time, we offer project follow-up, troubleshooting assistance, and Q&A sessions to address any queries related to the results
Sample Requirements and Delivery
|
Library |
Sequencing strategy |
Data recommended |
Quality Control |
|
rRNA depleted |
Illumina PE150 |
16 Gb |
Q30≥85% |
|
Size selected |
Illumina SE50 |
10-20M reads |
Sample Requirements:
Nucleotides:
|
Conc.(ng/μl) |
Amount (μg) |
Purity |
Integrity |
|
≥ 100 |
≥ 1 |
OD260/280=1.7-2.5 OD260/230=0.5-2.5 Limited or no protein or DNA contamination shown on gel. |
Plants: RIN≥6.5 Animal: RIN≥7.0 5.0≥28S/18S≥1.0; limited or no baseline elevation |
Recommended Sample Delivery
Container: 2 ml centrifuge tube (Tin foil is not recommended)
Sample labeling: Group+replicate e.g. A1, A2, A3; B1, B2, B3.
Shipment:
1. Dry-ice: Samples need to be packed in bags and buried in dry-ice.
2. RNAstable tubes: RNA samples can be dried in RNA stabilization tube(e.g. RNAstable®) and shipped at room temperature.
Service Work Flow

Experiment design

Sample delivery

RNA extraction

Library construction

Sequencing

Data analysis

After-sale services
Bioinformatics
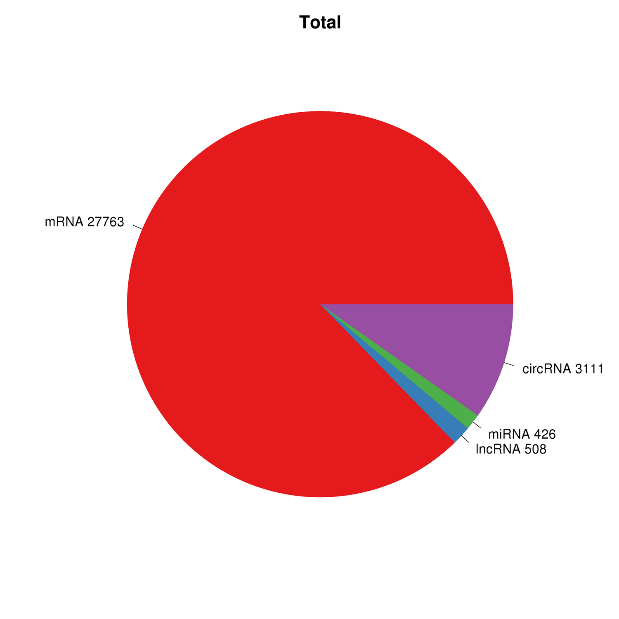
RNA expression overview
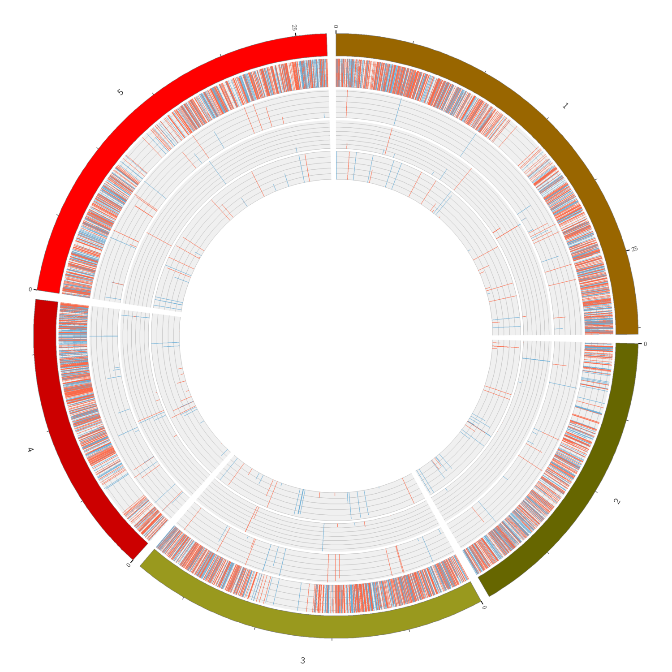
Differentially Expressed genes
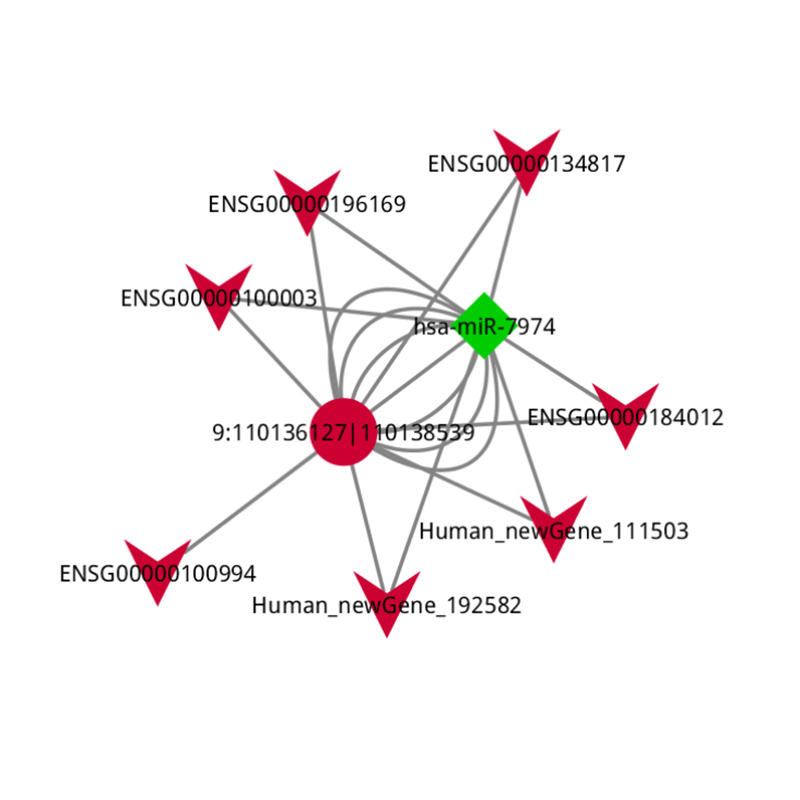
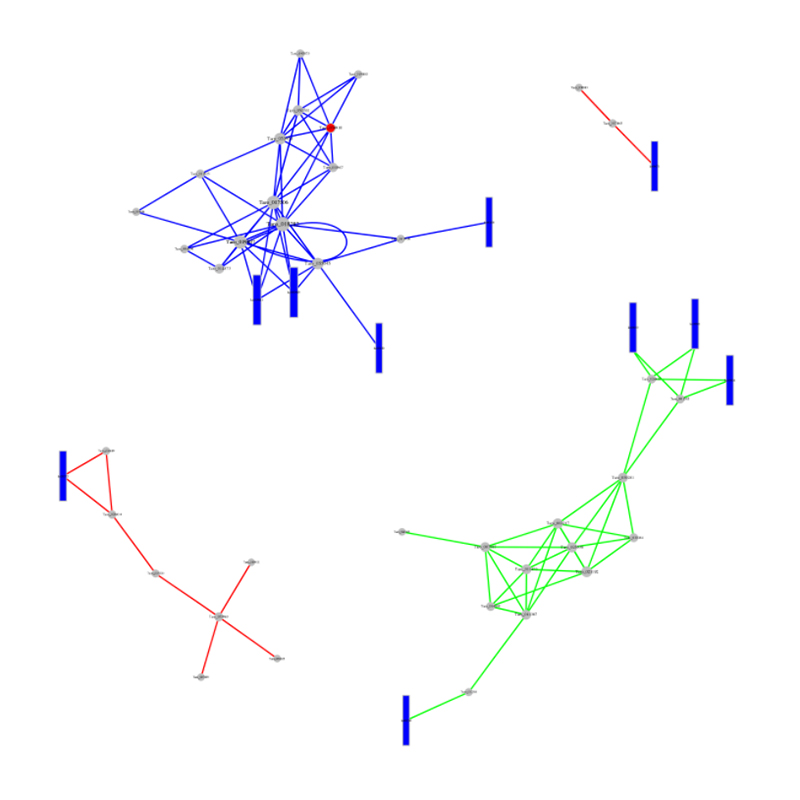
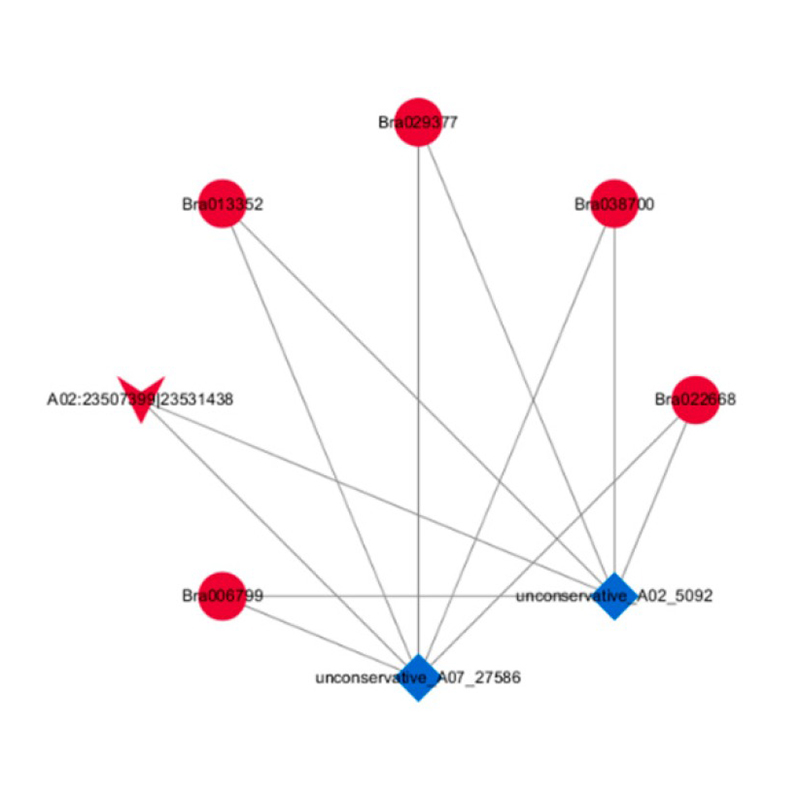
ceRNA analysis
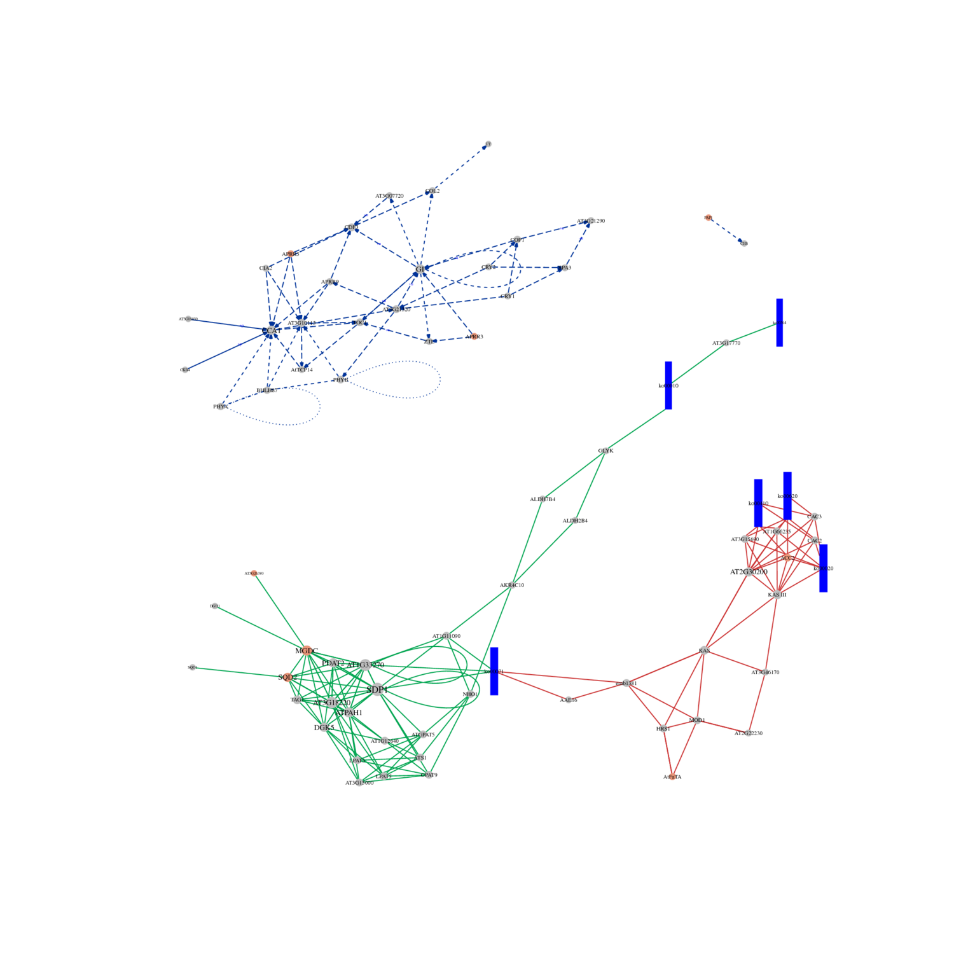 Differentially expressed miRNAs and related RNAs
Differentially expressed miRNAs and related RNAs
Explore the research advancements facilitated by BMKGene’ whole transcriptome sequencing services through a curated collection of publications.
Dai, Y. et al. (2022) ‘Comprehensive expression profiles of mRNAs, lncRNAs and miRNAs in Kashin-Beck disease identified by RNA-sequencing’, Molecular Omics, 18(2), pp. 154–166. doi: 10.1039/D1MO00370D.
Liu, N. nan et al. (2022) ‘Full length transcriptomes analysis of cold-resistance of Apis cerana in Changbai Mountain during overwintering period.’, Gene, 830, pp. 146503–146503. doi: 10.1016/J.GENE.2022.146503.
Wang, X. J. et al. (2022) ‘Multi-Omics Integration-Based Prioritisation of Competing Endogenous RNA Regulation Networks in Small Cell Lung Cancer: Molecular Characteristics and Drug Candidates’, Frontiers in Oncology, 12, p. 904865. doi: 10.3389/FONC.2022.904865/BIBTEX.
Xu, P. et al. (2022) ‘Integrated analysis of the lncRNA/circRNA-miRNA-mRNA expression profiles reveals novel insights into potential mechanisms in response to root-knot nematodes in peanut’, BMC Genomics, 23(1), pp. 1–12. doi: 10.1186/S12864-022-08470-3/FIGURES/7.
Yan, Z. et al. (2022) ‘Whole-transcriptome RNA sequencing highlights the molecular mechanisms associated with the maintenance of postharvest quality in broccoli by red LED irradiation’, Postharvest Biology and Technology, 188, p. 111878. doi: 10.1016/J.POSTHARVBIO.2022.111878.