Human Whole Exome Sequencing

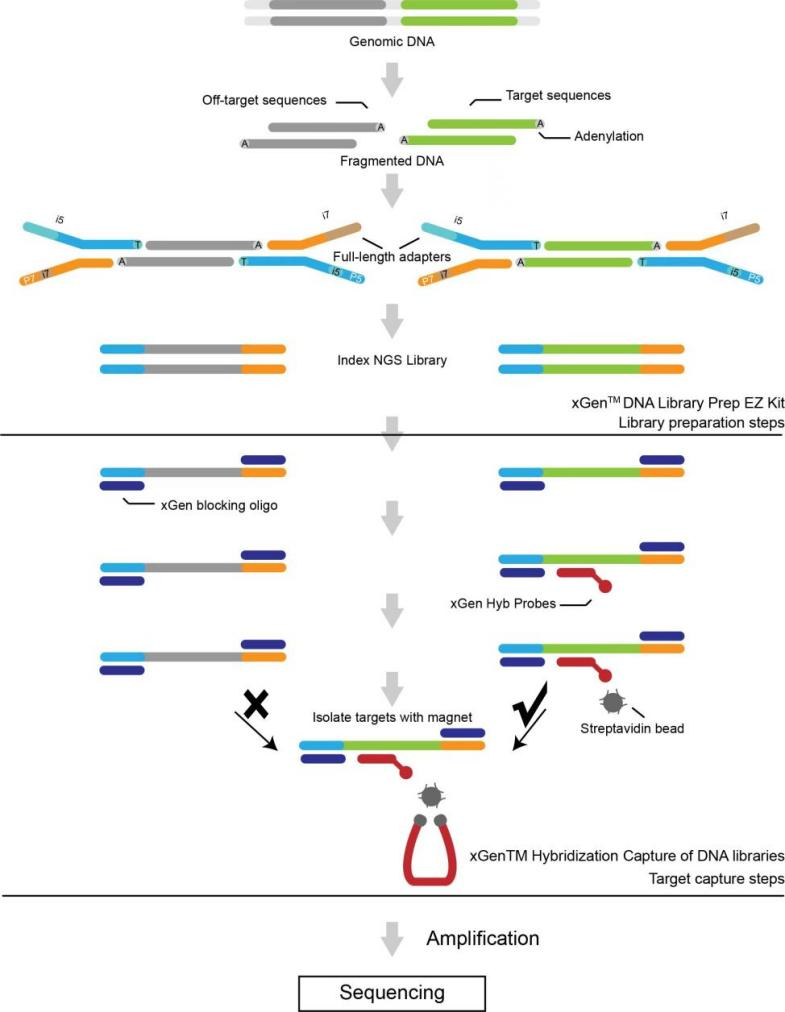
Service Features
● Two exome panels available based on target enrichment with probes: Sure Select Human All Exon v6 (Agilent) and xGen Exome Hybridization Panel v2 (IDT).
● Sequencing on Illumina NovaSeq.
● Bioinformatic pipeline directed towards disease analysis or tumor analysis.
Service Advantages
● Targets Protein Coding Region: By capturing and sequencing protein coding regions, hWES is utilized to reveal variants related to protein structure.
● Cost Effective: hWES yields approximately 85% of the human disease-associated mutations from 1% of the human genome.
● High Accuracy: With high sequencing depth, hWES facilitates the detection of both common variants and rare variants with frequencies lower than 1%.
● Rigorous Quality Control: We implement five core control points across all stages, from sample and library preparation to sequencing and bioinformatics. This meticulous monitoring ensures the delivery of consistently high-quality results.
● Comprehensive bioinformatics analysis: our pipeline goes beyond identifying variations to the reference genome, as it incorporates advanced methods designed to specifically address research questions related to genetic aspects of diseases or tumor analysis.
● Post-Sales Support: Our commitment extends beyond project completion with a 3-month after-sale service period. During this time, we offer project follow-up, troubleshooting assistance, and Q&A sessions to address any queries related to the results.
Sample Specifications
|
Exon capture Strategy |
Sequencing Strategy |
Recommended data output |
Quality control |
|
Sure Select Human All Exon v6 (Agilent) or xGen Exome Hybridization Panel v2 (IDT)
|
Illumina NovaSeq PE150 |
5 -10 Gb For mendelian disorders/rare diseases: > 50x For tumor samples: < 100x |
Q30≥85% |
Sample Requirements
|
Sample Type
|
Amount (Qubit® )
|
Volume
|
Concentration
|
Purity(NanoDrop™ ) |
|
Genomic DNA
|
≥ 300 ng |
≥ 15 μL
|
≥ 20 ng/μL
|
OD260/280=1.8-2.0
no degradation, no contamination
|
Recommended Sequencing Depth
For Mendelian disorders/rare diseases: effective sequencing depth above 50×
For tumor samples: effective sequencing depth above 100×
Bioinformatics
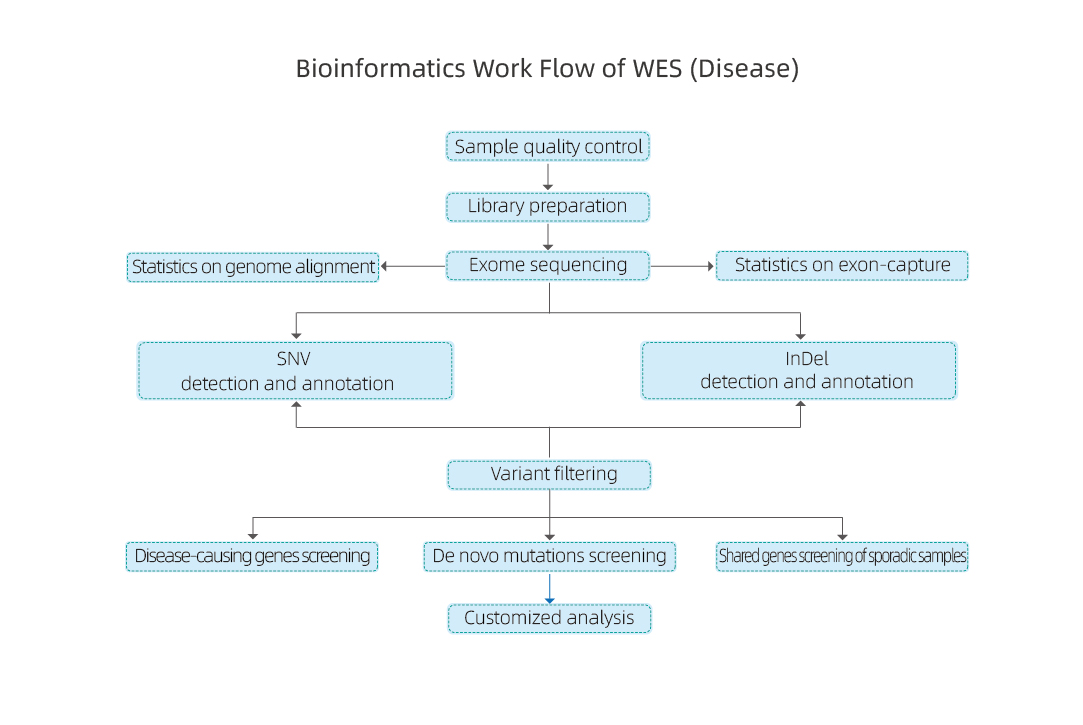
Bioinformatic analysis of hWEs-disease includes:
● Sequencing data QC
● Reference Genome Alignment
● Identification of SNPs and InDels
● Functional Annotation of SNPs and InDels
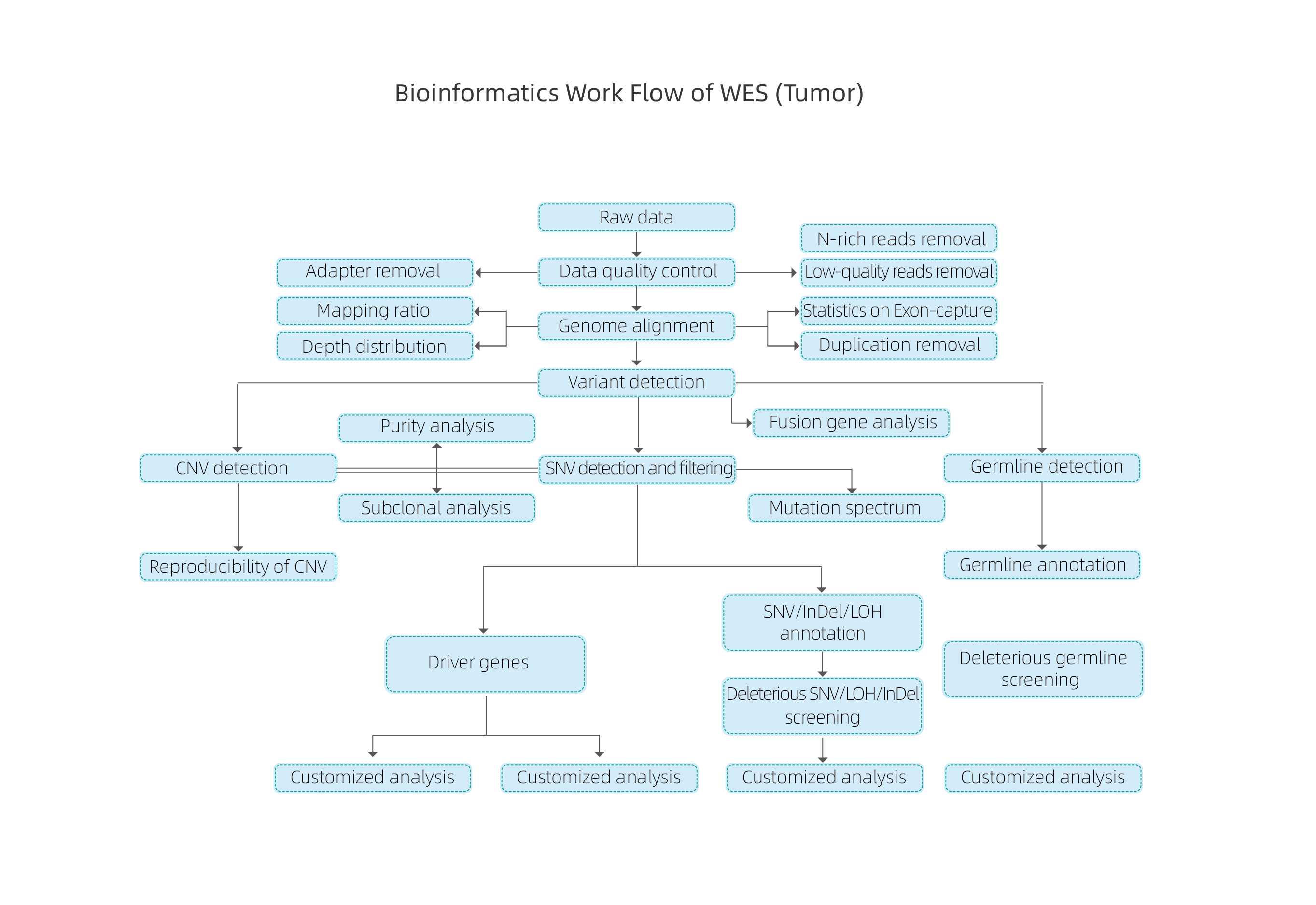
Bioinformatic analysis of tumor samples includes:
● Sequencing data QC
● Reference Genome Alignment
● Identification of SNPs, InDels and somatic variations
● Identification of germline variants
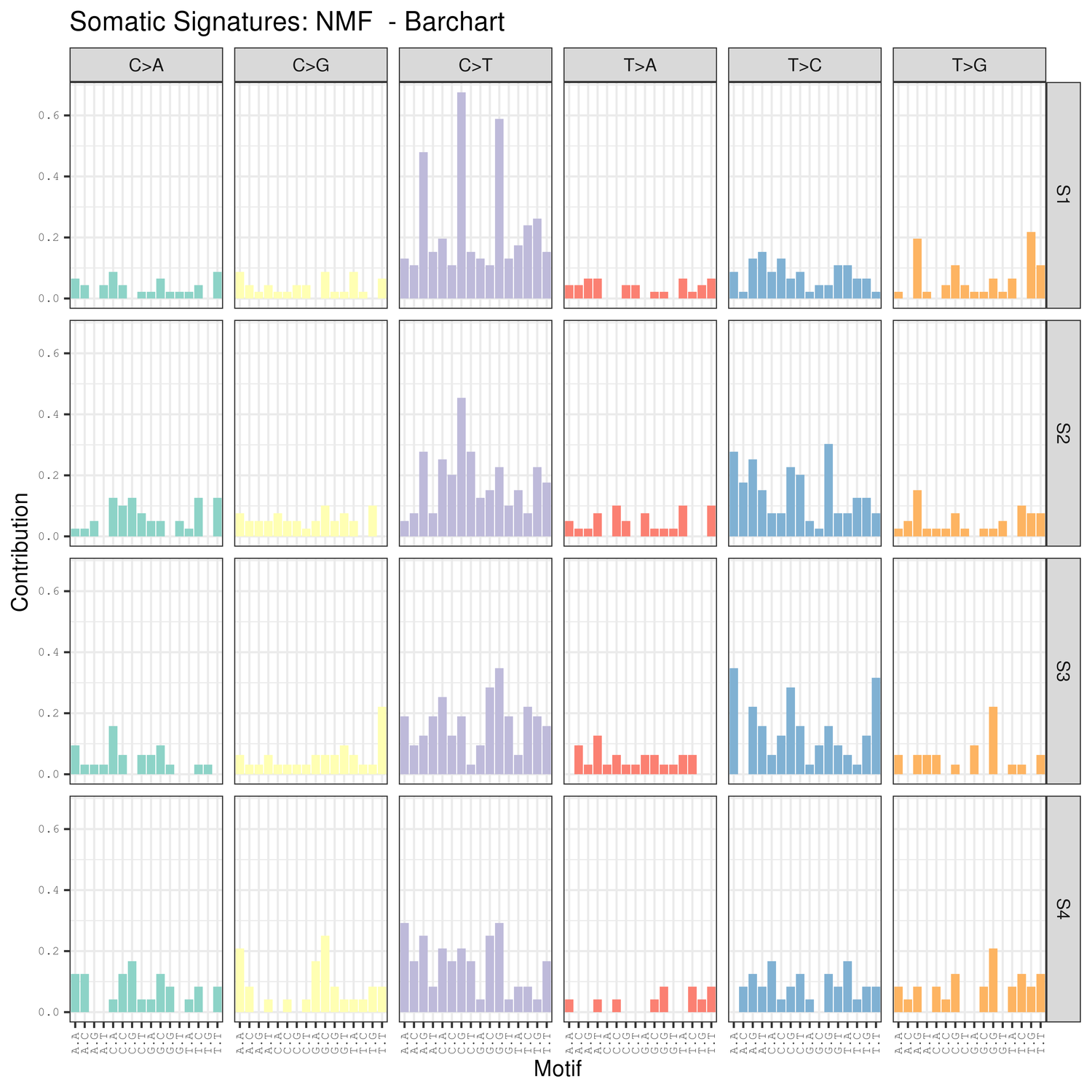
● Mutation signatures analysis
● Identification of drive genes based on gain-of-function mutations
● Mutation annotation at the level of drug susceptibility
● Heterogeneity analysis – calculation of purity and ploidy
Service Work Flow
Sample delivery

DNA extraction

Library construction

Sequencing

Data analysis
Data delivery

After-sale services
Data QC – Statistics of Exome capture
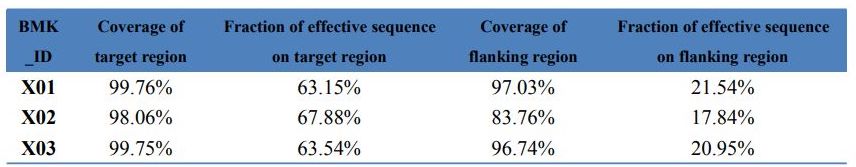
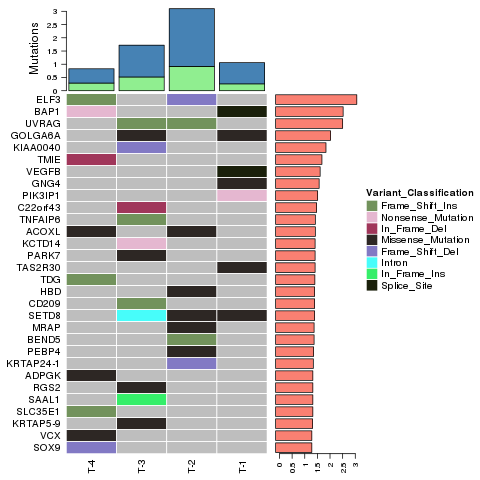
Variant identification – InDels
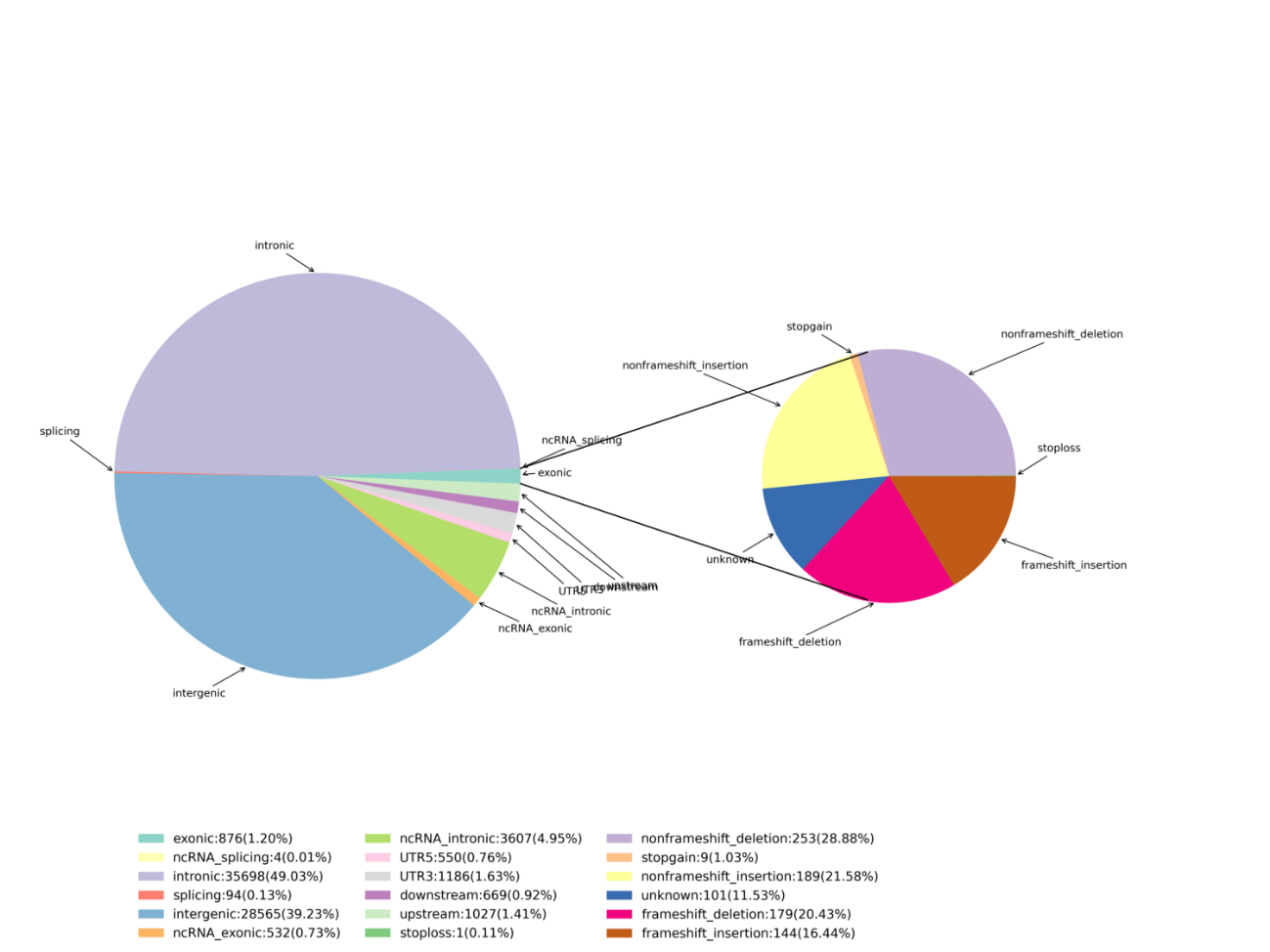
Advanced analysis: identification and distribution of deleterious SNPs/InDels – Circos plot

Tumor analysis: identification and distribution of somatic mutations – Circos plot
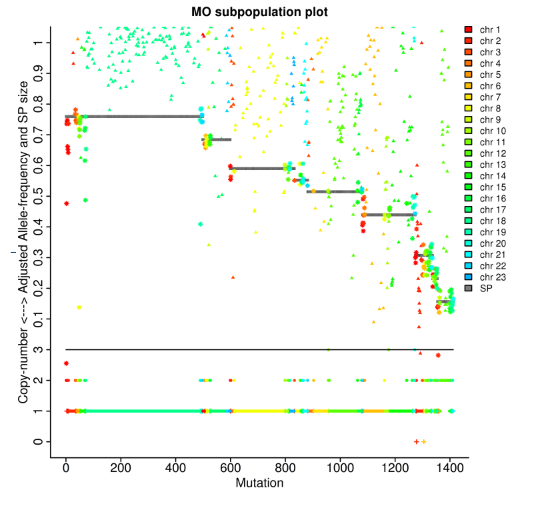
Tumor analysis: clonal lineages