

Specific-Locus Amplified Fragment Sequencing (SLAF-Seq)

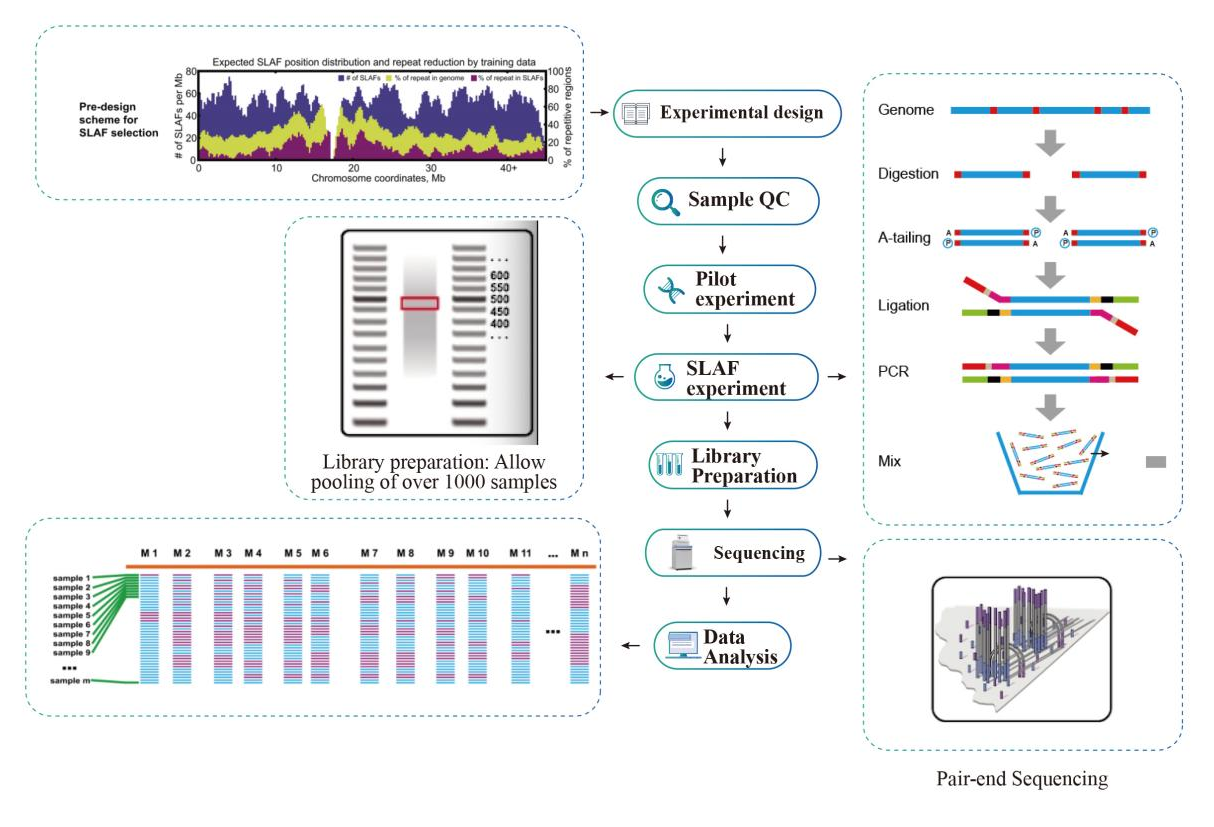
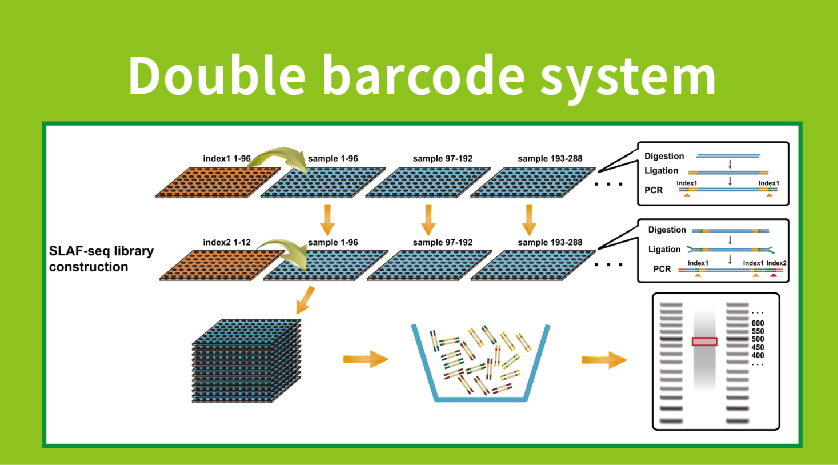
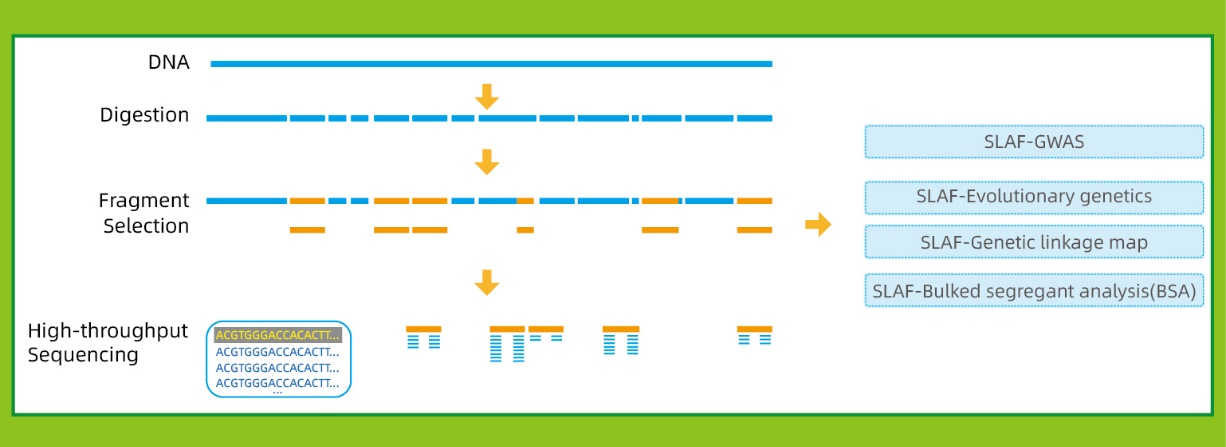
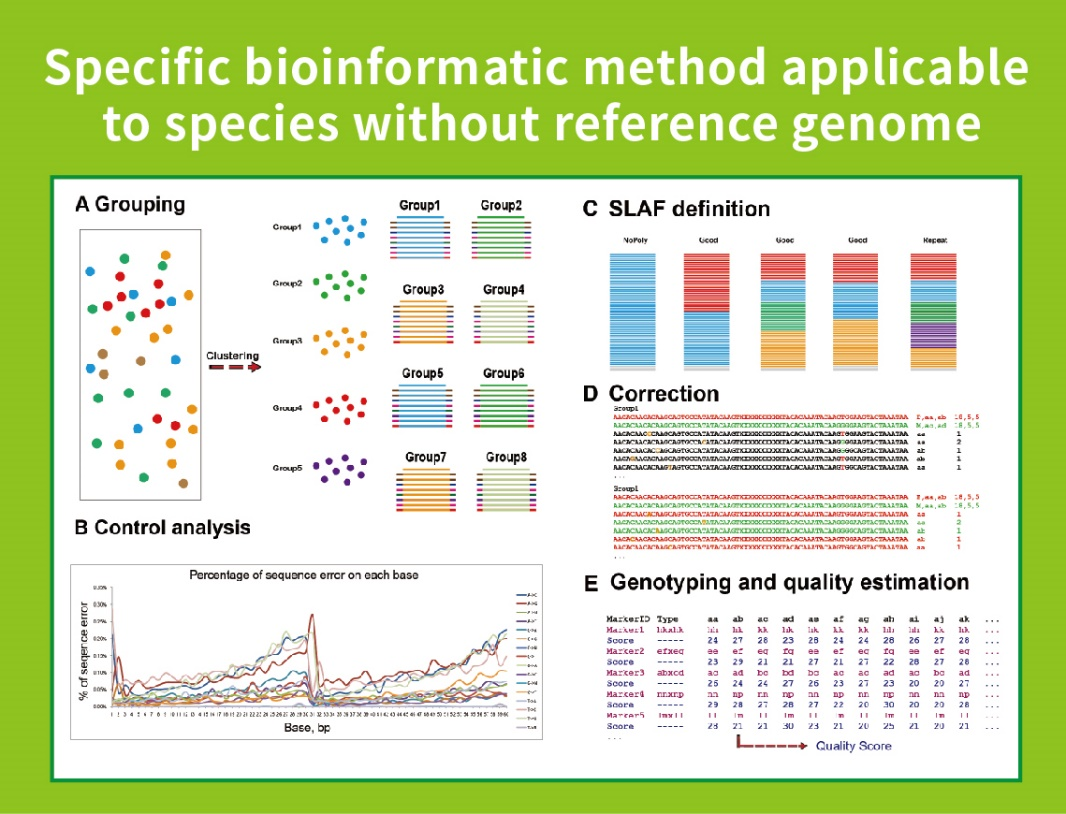
Workflow

Technical Scheme

Service Features
● Sequencing on NovaSeq with PE150.
● Library preparation with double barcoding, enabling pooling of over 1000 samples.
● This technique can be used with or without a reference genome, with different bioinformatic pipelines for each case:
With reference genome: SNP and InDel discovery
Without reference genome: sample clustering and SNP discovery
● In the in silico pre-design stage multiple restriction enzyme combinations are screened to find the ones that generate a uniform distribution of SLAF tags along the genome.
● During the pre-experiment, three enzyme combinations are tested in 3 samples to generate 9 SLAF libraries, and this information is use to choose the optimal restriction enzyme combination for the project.
Service Advantages
● High Genetic Marker Discovery: Integrating a high-throughput double barcode system allows for the simultaneous sequencing of large populations, and locus-specific amplification enhances efficiency, ensuring that tag numbers meet the diverse requirements of various research questions.
● Low Dependence on the Genome: It can be applied to species with or without a reference genome.
● Flexible Scheme Design: Single-enzyme, dual-enzyme, multi-enzyme digestion, and various types of enzymes can all be selected to cater to different research goals or species. The in silico pre-design is carried out to ensure an optimal enzyme design.
● High Efficiency in Enzymatic Digestion: The conduction of an in silico pre-design and a pre-experiment assured optimal design with even distribution of SLAF tags on the chromosome (1 SLAF tag/4Kb) and reduced repetitive sequence (<5%).
● Extensive Expertise: Our team brings a wealth of experience to every project, with a track record of closing over 2000 SLAF-Seq projects on hundreds of species, including plants, mammals, birds, insects, and aquatic organisms.
● Self-developed Bioinformatic Workflow: BMKGENE developed an integrated bioinformatic workflow for SLAF-Seq to ensure the reliability and accuracy of the final output.
Service Specifications
|
Type of analysis |
Recommended population scale |
Sequencing strategy |
|
|
Depth of tag sequencing |
Tag number |
||
|
Genetic Maps |
2 parents and >150 offspring |
Parents: 20x Offsping: 10x |
Genome size: <400 Mb: WGS is recommended <1Gb: 100K tags 1-2Gb:: 200K tags >2Gb: 300K tags Max 500k tags |
|
Genome-Wide Association Studies (GWAS) |
200 samples |
10x |
|
|
Genetic Evolution |
30 samples, with >10 samples from each subgroup |
10x |
|
Service requirements
Concentration ≥ 5 ng/µL
Total amount ≥80 ng
Nanodrop OD260/280=1.6-2.5
Agarose gel: no or limited degradation or contamination
Recommended Sequencing Strategy
Container:
2 ml centrifuge tube
For most of the samples, we recommend not to preserve in ethanol.
Sample labeling: Samples need to be clearly labeled and identical to submitted sample information form.
Shipment:
Dry-ice: Samples need to be packed in bags first and buried in dry-ice.
Service Workflow






Sample QC
Pilot experiment
SLAF-experiment
Library Preparation
Sequencing
Data Analysis
After-sale Services
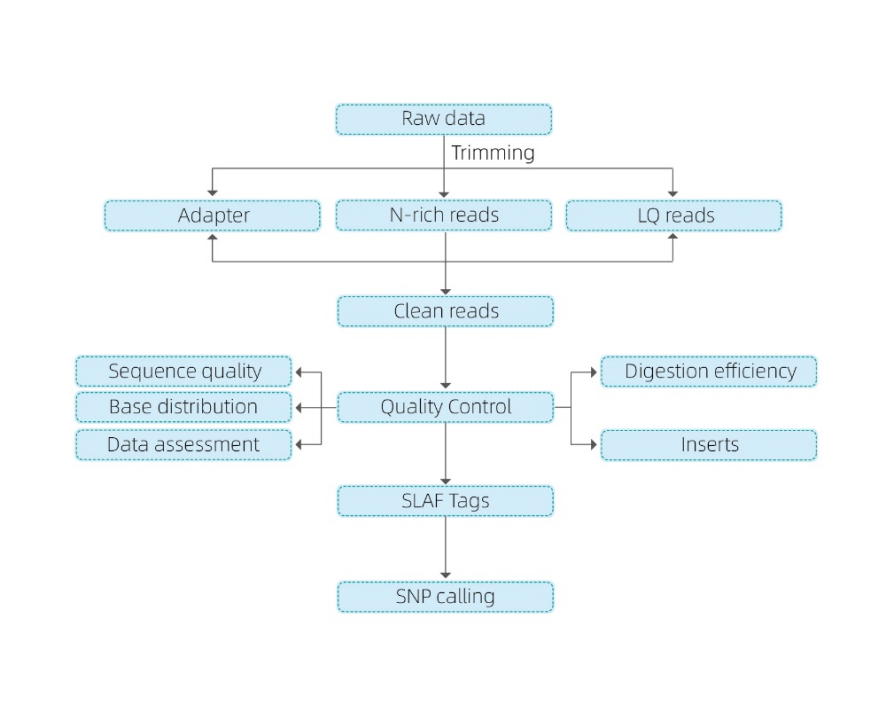 Includes the following analysis:
Includes the following analysis:
- Sequencing data QC
- SLAF tag development
Mapping to reference genome
Without a reference genome: clustering
- Analysis of SLAF tags.: statistics, distribution across the genome
- Marker discovery: SNP, InDel, SNV, CV calling and annotation
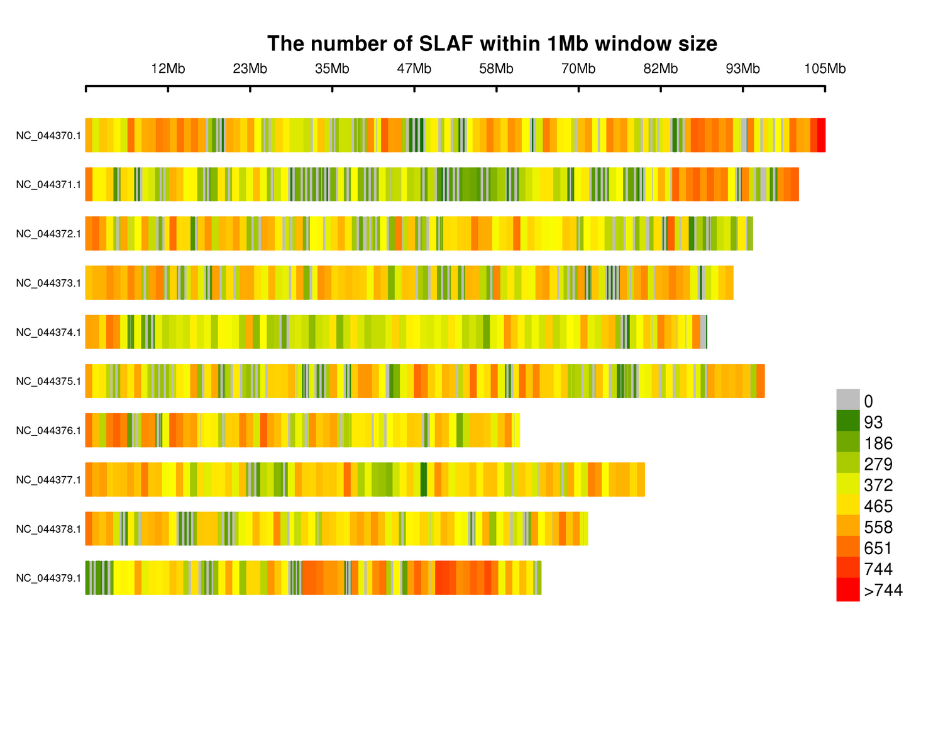
Distribution of SLAF tags on chromosomes:
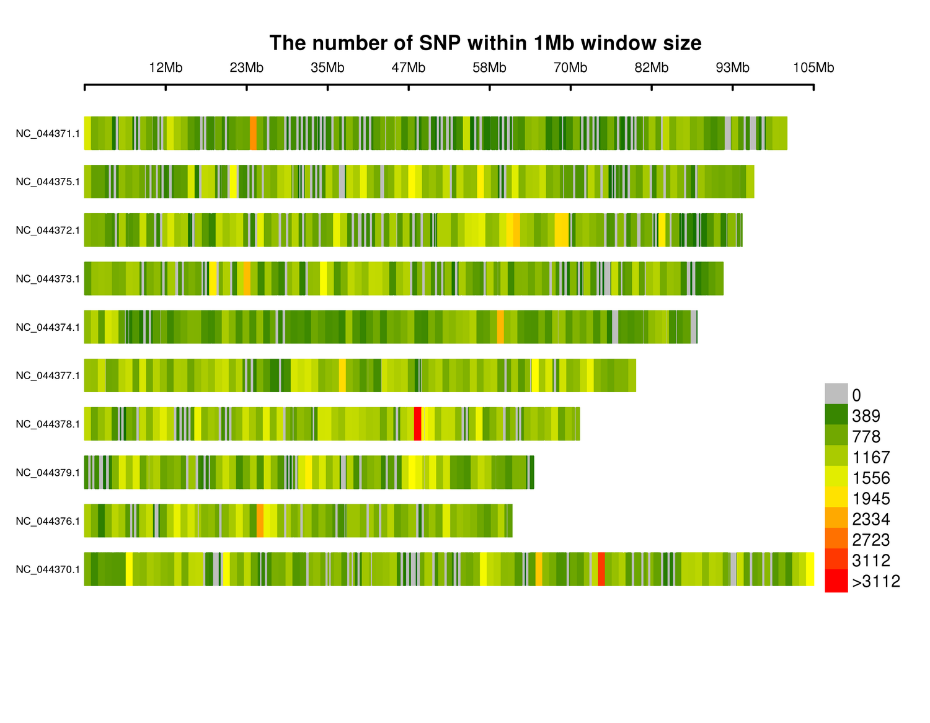
Distribution of SNPs on chromosomes:
 SNP annotation
SNP annotation
|
Year |
Journal |
IF |
Title |
Applications |
|
2022 |
Nature communications |
17.694 |
Genomic basis of the giga-chromosomes and giga-genome of tree peony Paeonia ostii |
SLAF-GWAS |
|
2015 |
New Phytologist |
7.433 |
Domestication footprints anchor genomic regions of agronomic importance in soybeans |
SLAF-GWAS |
|
2022 |
Journal of Advanced Research |
12.822 |
Genome-wide artificial introgressions of Gossypium barbadense into G. hirsutum reveal superior loci for simultaneous improvement of cotton fiber quality and yield traits |
SLAF-Evolutionary genetics |
|
2019 |
Molecular Plant |
10.81 |
Population Genomic Analysis and De Novo Assembly Reveal the Origin of Weedy Rice as an Evolutionary Game |
SLAF-Evolutionary genetics |
|
2019 |
Nature Genetics |
31.616 |
Genome sequence and genetic diversity of the common carp, Cyprinus carpio |
SLAF-Linkage map |
|
2014 |
Nature Genetics |
25.455 |
The genome of cultivated peanut provides insight into legume karyotypes, polyploid evolution and crop domestication. |
SLAF-Linkage map |
|
2022 |
Plant Biotechnology Journal |
9.803 |
Identification of ST1 reveals a selection involving hitchhiking of seed morphology and oil content during soybean domestication |
SLAF-Marker development |
|
2022 |
International Journal of Molecular Sciences |
6.208 |
Identification and DNA Marker Development for a Wheat-Leymus mollis 2Ns (2D) Disomic Chromosome Substitution |
SLAF-Marker development |
|
Year |
Journal |
IF |
Title |
Applications |
|
2023 |
Frontiers in plant science |
6.735 |
QTL mapping and transcriptome analysis of sugar content during fruit ripening of Pyrus pyrifolia |
Genetic Map |
|
2022 |
Plant Biotechnology Journal |
8.154 |
Identification of ST1 reveals a selection involving hitchhiking of seed morphology and oil content during soybean domestication
|
SNP calling |
|
2022 |
Frontiers in plant science |
6.623 |
Genome-Wide Association Mapping of Hulless Barely Phenotypes in Drought Environment.
|
GWAS |





