Full-Length mRNA Sequencing-Nanopore

Features
● Capture of poly-A mRNA followed by cDNA synthesis and library preparation
● Sequencing of the full-length transcripts
● Bioinformatic analysis based on alignment to a reference genome
● Bioinformatic analysis includes not only expression at gene and isoform-level but also analysis of lncRNA, gene fusions, poly-adenylation and gene structure
Service Advantages
● Quantification of expression at the isoform level: enabling detailed and accurate expression analysis, unveiling change that may be masked when analysing the whole gene expression
● Reduced Data Demands: Compared to Next-Generation Sequencing (NGS), Nanopore sequencing exhibits lower data requirements, allowing for equivalent levels of gene expression quantification saturation with smaller data.
● Higher accuracy of expression quantification: both at gene and isoform level
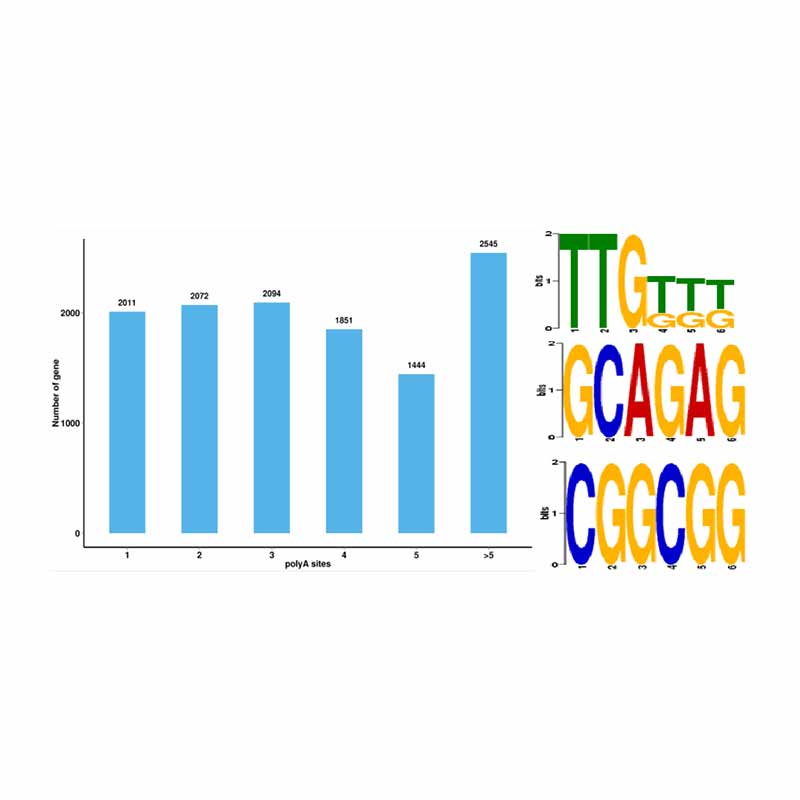
● Identification of additional transcriptomic information: alternative polyadenylation, fusion genes and lcnRNA and their target genes
● Extensive Expertise: Our team brings a wealth of experience to every project, having completed over 550 Nanopore full-length transcriptome projects and processed over 7,700 samples.
● Post-Sales Support: our commitment extends beyond project completion with a 3-month after-sale service period. During this time, we offer project follow-up, troubleshooting assistance, and Q&A sessions to address any queries related to the results.
Sample Requirements and Delivery
|
Library |
Sequencing strategy |
Data recommended |
Quality Control |
|
Poly A enriched |
Illumina PE150 |
6 Gb |
Average quality score: Q10 |
Sample Requirements:
Nucleotides:
|
Conc.(ng/μl) |
Amount (μg) |
Purity |
Integrity |
|
≥ 100 |
≥ 0.6 |
OD260/280=1.7-2.5 OD260/230=0.5-2.5 Limited or no protein or DNA contamination shown on gel. |
For plants: RIN≥7.5; For animals: RIN≥7.0; 5.0≥28S/18S≥1.0; limited or no baseline elevation |
● Plants:
Root, Stem or Petal: 450 mg
Leaf or Seed: 300 mg
Fruit: 1.2 g
● Animal:
HEart or Intestine: 300 mg
Viscera or Brain: 240 mg
Muscle: 450 mg
Bones, Hair or Skin: 1g
● Arthropods:
Insects: 6g
Crustacea: 300 mg
● Whole blood: 1 tube
● Cells: 106 cells
Recommended Sample Delivery
Container: 2 ml centrifuge tube (Tin foil is not recommended)
Sample labeling: Group+replicate e.g. A1, A2, A3; B1, B2, B3.
Shipment:
1. Dry-ice: Samples need to be packed in bags and buried in dry-ice.
2. RNAstable tubes: RNA samples can be dried in RNA stabilization tube(e.g. RNAstable®) and shipped in room temperature.
Service Work Flow
Nucleotides:

Sample delivery

Library construction

Sequencing

Data analysis

After-sale services
Service Work Flow
Tissue:

Experiment design
Sample delivery

RNA extraction
Library construction
Sequencing
Data analysis
After-sale services
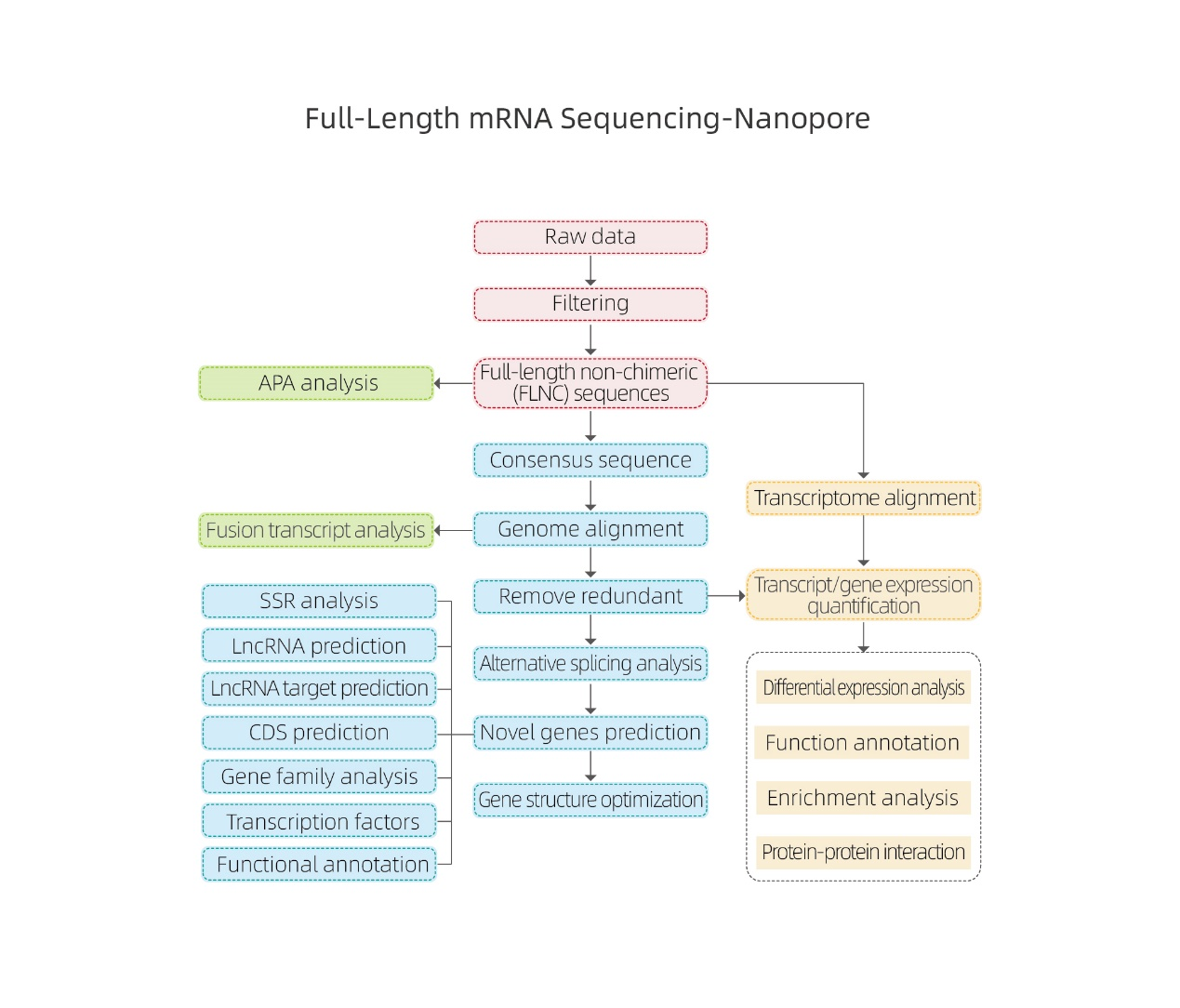
● Raw data processing
● Transcript identification
● Alternative splicing
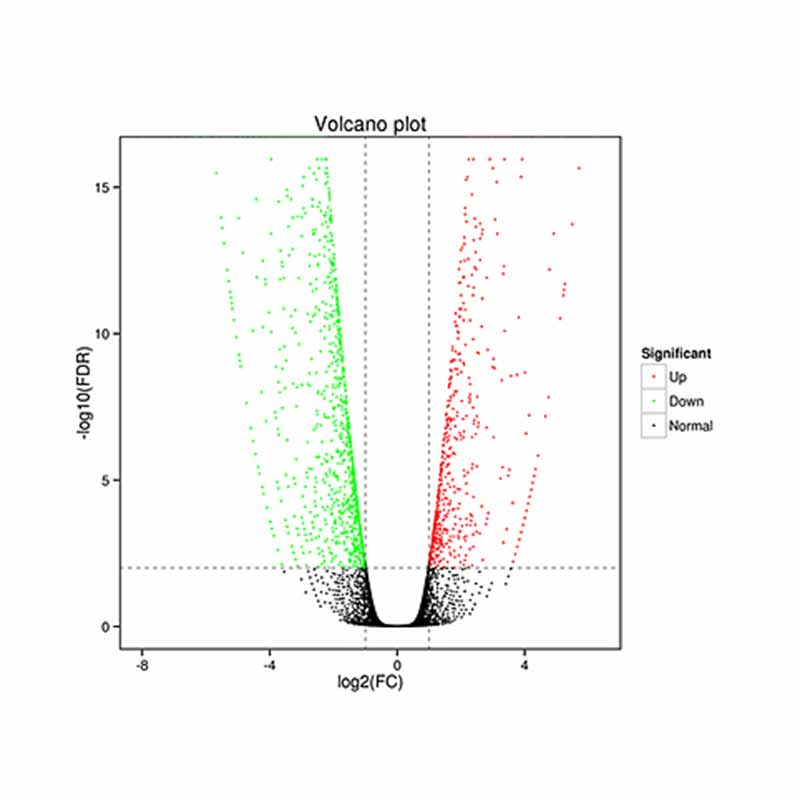
● Expression quantification in gene level and isoform level
● Differential expression analysis
● Function annotation and enrichment (DEGs and DETs)
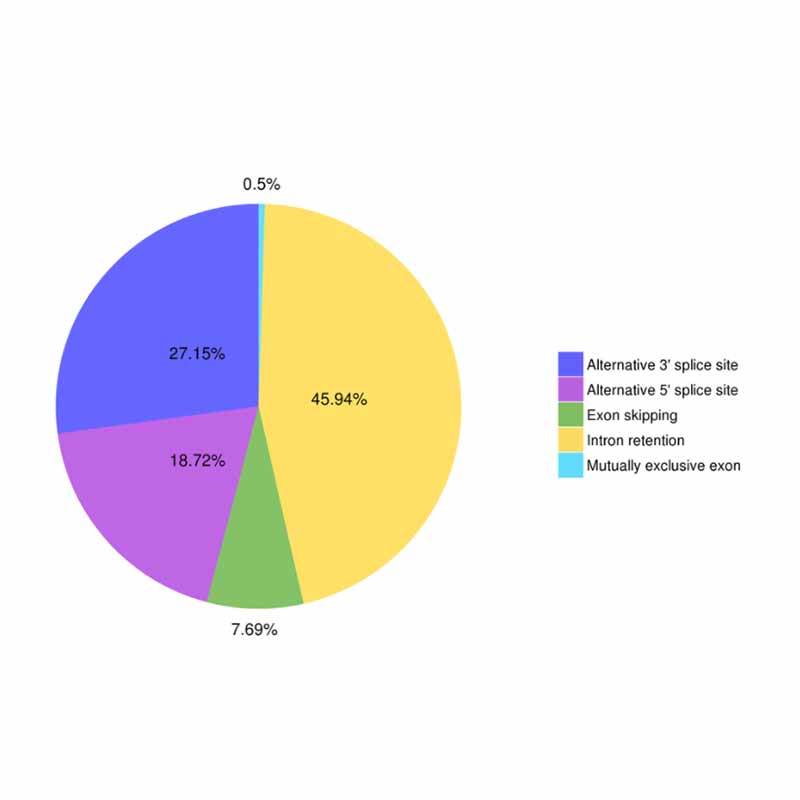
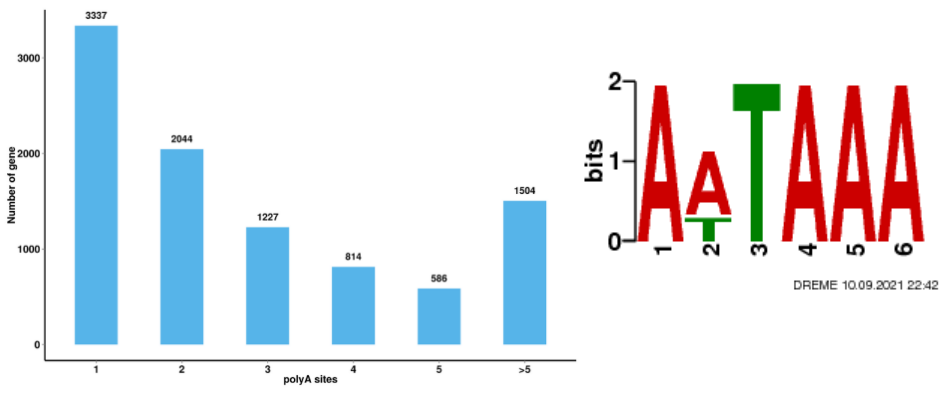
Alternative splicing analysis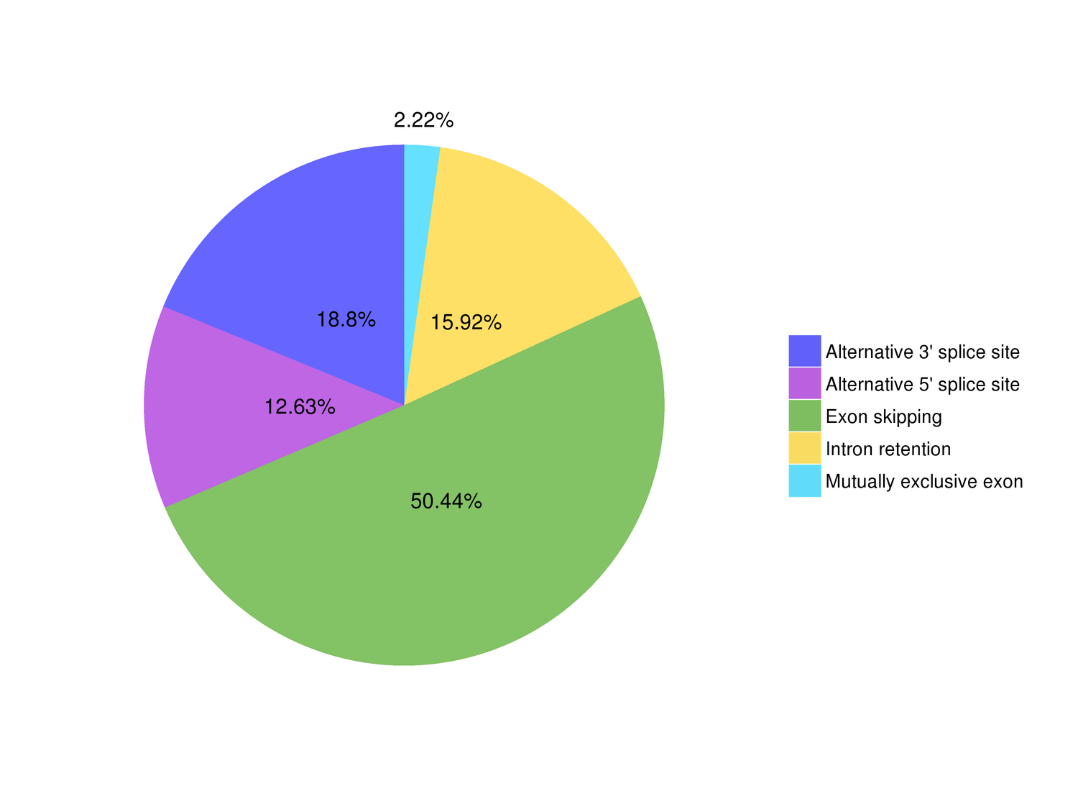 Alternative Polyadenylation Analysis (APA)
Alternative Polyadenylation Analysis (APA)
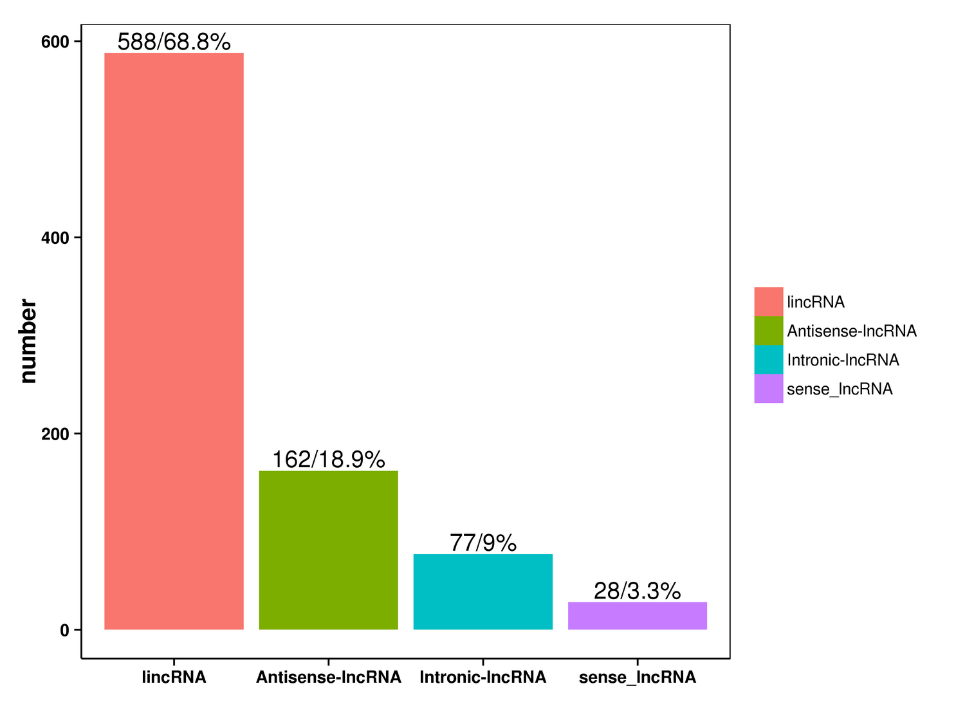
lncRNA prediction
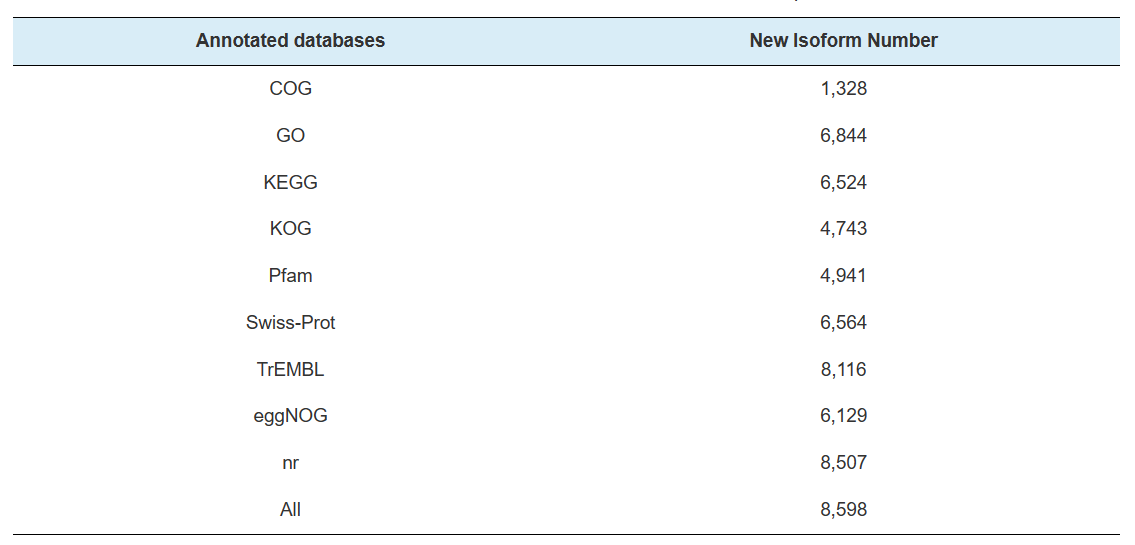
Annotation of novel genes
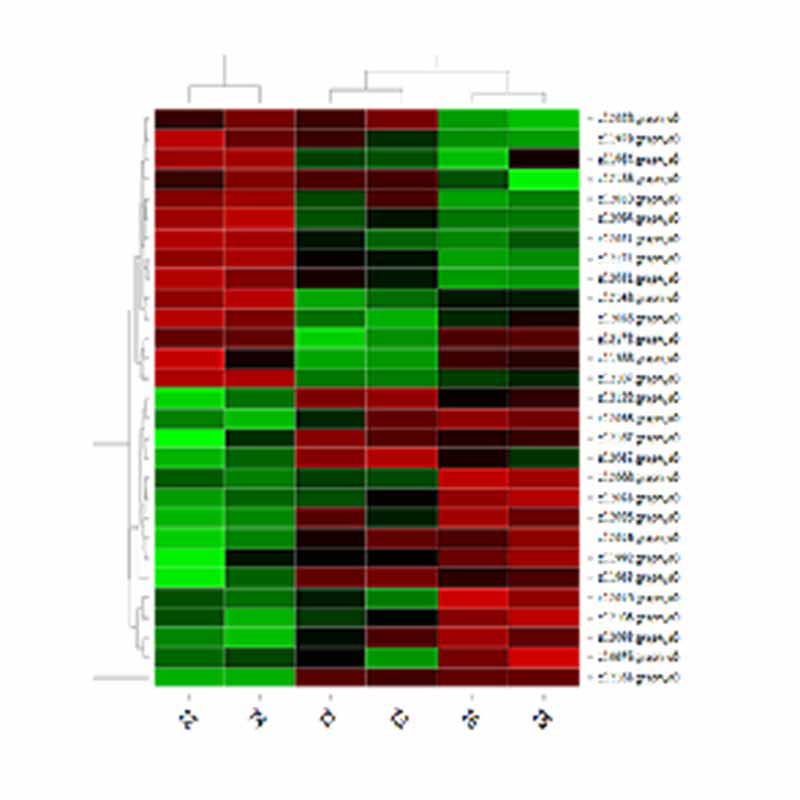
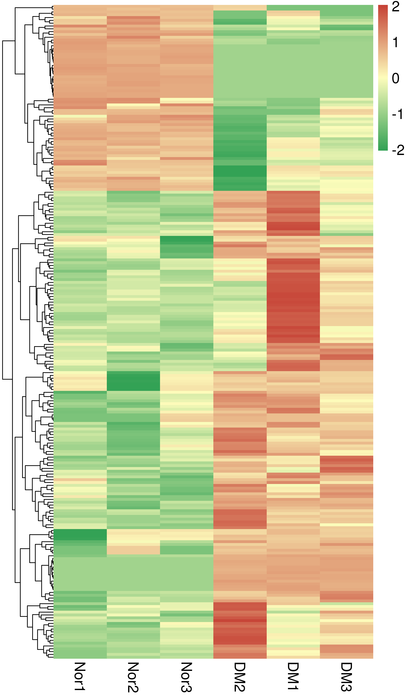
Clustering of DETs
Protein-Protein Networks in DEGs
Explore the advancements facilitated by BMKGene’s Nanopore full-length mRNA sequencing services through a curated collection of publications.
Gong, B. et al. (2023) ‘Epigenetic and transcriptional activation of the secretory kinase FAM20C as an oncogene in glioma’, Journal of Genetics and Genomics, 50(6), pp. 422–433. doi: 10.1016/J.JGG.2023.01.008.
He, Z. et al. (2023) ‘Full-length transcriptome sequencing of lymphocytes respond to IFN-γ reveals a Th1-skewed immune response in flounder (Paralichthys olivaceus)’, Fish & Shellfish Immunology, 134, p. 108636. doi: 10.1016/J.FSI.2023.108636.
Ma, Y. et al. (2023) ‘Comparative analysis of PacBio and ONT RNA sequencing methods for Nemopilema Nomurai venom identification’, Genomics, 115(6), p. 110709. doi: 10.1016/J.YGENO.2023.110709.
Yu, D. et al. (2023) ‘Nano-seq analysis reveals different functional tendency between exosomes and microvesicles derived from hUMSC’, Stem Cell Research and Therapy, 14(1), pp. 1–13. doi: 10.1186/S13287-023-03491-5/TABLES/6.