

De-novo-Genomsequenzierung von Pflanzen und Tieren




Servicevorteile
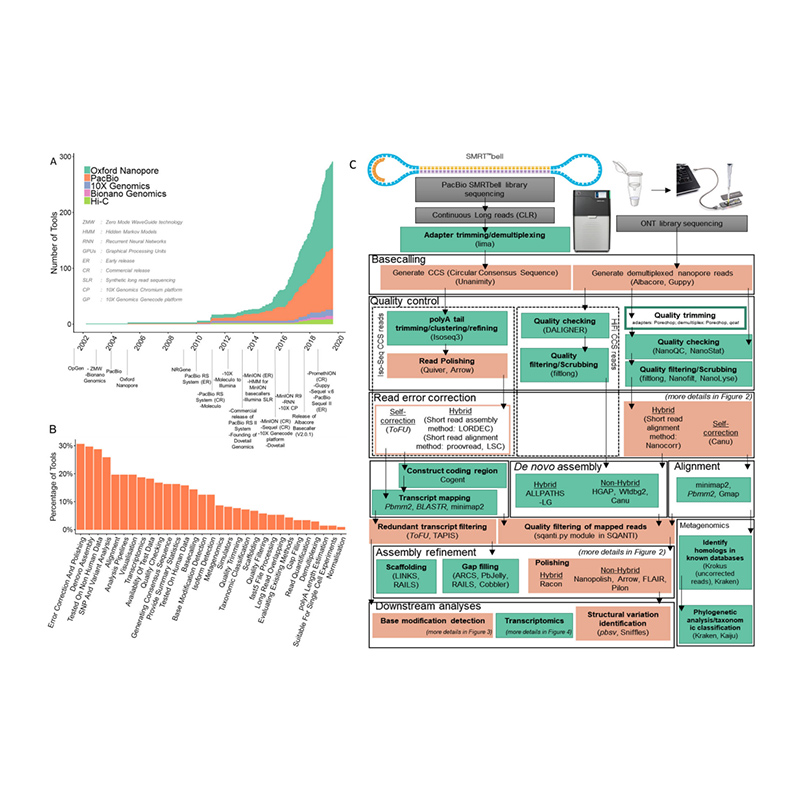
Entwicklung von Sequenzierungsplattformen und Bioinformatik inde novoGenomassemblierung
(Amarasinghe SL et al.,Genombiologie, 2020)
● Konstruktion neuer Genome und Verbesserung bestehender Referenzgenome für interessierende Arten.
● Höhere Genauigkeit, Kontinuität und Vollständigkeit bei der Montage
● Aufbau grundlegender Ressourcen für die Forschung in den Bereichen Sequenzpolymorphismus, QTLs, Genbearbeitung, Züchtung usw.
● Ausgestattet mit dem gesamten Spektrum an Sequenzierungsplattformen der dritten Generation: Komplettlösung für die Genomassemblierung
● Flexible Sequenzierungs- und Assemblierungsstrategien, die verschiedene Genome mit unterschiedlichen Merkmalen erfüllen
● Hochqualifiziertes Bioinformatiker-Team mit großer Erfahrung in komplexen Genomassemblierungen, einschließlich Polyploiden, Riesengenomen usw.
● Über 100 erfolgreiche Fälle mit einem kumulativen veröffentlichten Impact Factor von über 900
● Bearbeitungszeit von nur 3 Monaten für die Genomassemblierung auf Chromosomenebene.
● Solide technische Unterstützung mit einer Reihe von Patenten und Software-Urheberrechten sowohl im experimentellen Bereich als auch in der Bioinformatik.
Leistungsbeschreibung
|
Inhalt
|
Plattform
|
Leselänge
|
Abdeckung
|
| Genomuntersuchung
| Illumina NovaSeq
| PE150
| ≥ 50X
|
| Genomsequenzierung
| PacBio Revio
| 15 kb HiFi-Lesevorgänge
| ≥ 30X
|
| Hi-C
| Illumina NovaSeq
| PE150
| ≥100X
|
Arbeitsablauf

Musteranforderungen und Lieferung
Probenanforderungen:
| Spezies | Gewebe | Für PacBio | Für Nanopore |
| Tiere | Viszerale Organe (Leber, Milz usw.) | ≥ 1,0 g | ≥ 3,5 g |
| Muskel | ≥ 1,5 g | ≥ 5,0 g | |
| Blut von Säugetieren | ≥ 1,5 ml | ≥ 5,0 ml | |
| Blut von Fischen oder Vögeln | ≥ 0,2 ml | ≥ 0,5 ml | |
| Pflanzen | Frische Blätter | ≥ 1,5 g | ≥ 5,0 g |
| Blütenblatt oder Stiel | ≥ 3,5 g | ≥ 10,0 g | |
| Wurzeln oder Samen | ≥ 7,0 g | ≥ 20,0 g | |
| Zellen | Zellkultur | ≥ 3×107 | ≥ 1×108 |
Empfohlene Musterlieferung
Behälter: 2 ml Zentrifugenröhrchen (Alufolie wird nicht empfohlen)
Für die meisten Proben empfehlen wir, sie nicht in Ethanol aufzubewahren.
Probenkennzeichnung: Proben müssen deutlich gekennzeichnet sein und mit dem eingereichten Probeninformationsformular identisch sein.
Versand: Trockeneis: Die Proben müssen zunächst in Beutel verpackt und in Trockeneis vergraben werden.
Service-Workflow

Experimentdesign

Musterlieferung

DNA-Extraktion

Bibliotheksbau

Sequenzierung

Datenanalyse

Kundendienst
*Die hier gezeigten Demo-Ergebnisse stammen alle von Genomen, die mit Biomarker Technologies veröffentlicht wurden
1.Circos auf Chromosomenebene GenomassemblierungG. rotundifoliumvon der Nanopore-Sequenzierungsplattform
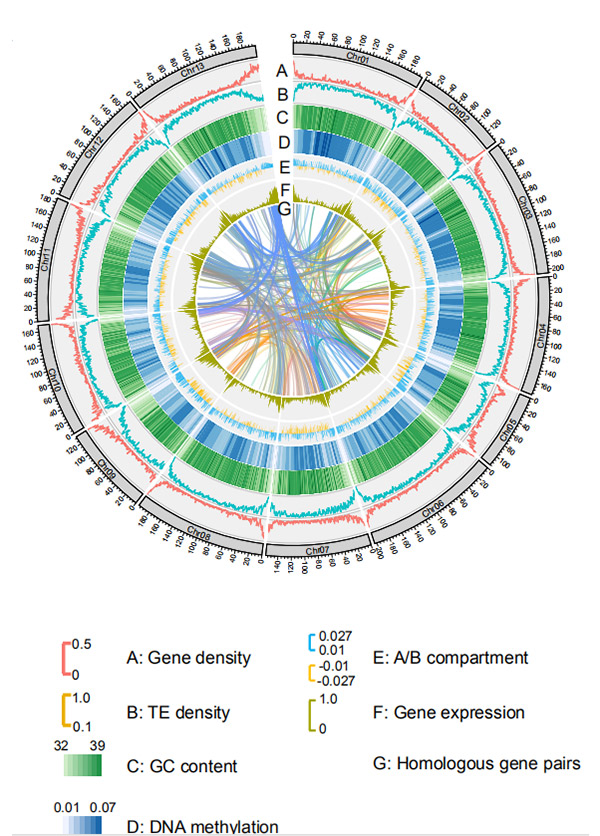
Wang M et al.,Molekularbiologie und Evolution, 2021
2.Statistik der Assemblierung und Annotation des Weining-Roggengenoms
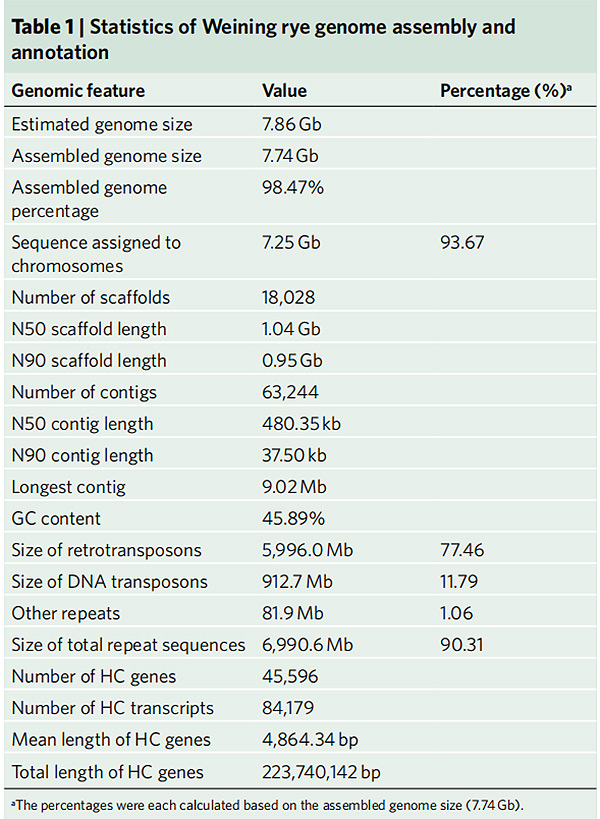
Li G et al.,Naturgenetik, 2021
3.Genvorhersage vonSechium eduleGenom, abgeleitet aus drei Vorhersagemethoden:De novoVorhersage, Homologie-basierte Vorhersage und RNA-Seq-Daten-basierte Vorhersage
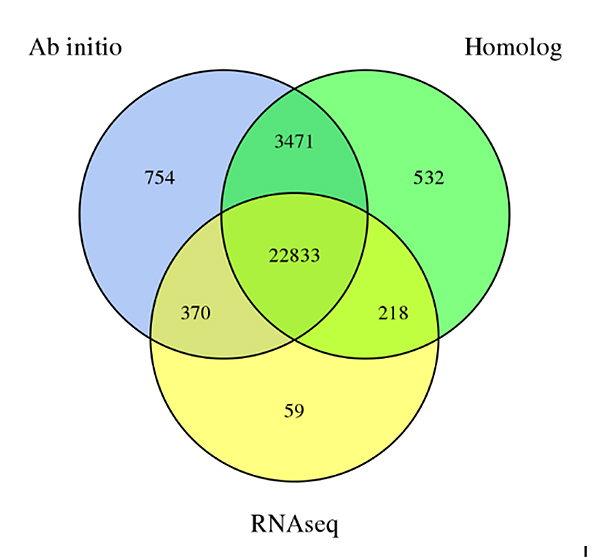
Fu A et al.,Gartenbauforschung, 2021
4.Identifizierung intakter langer terminaler Wiederholungen in drei Baumwollgenomen
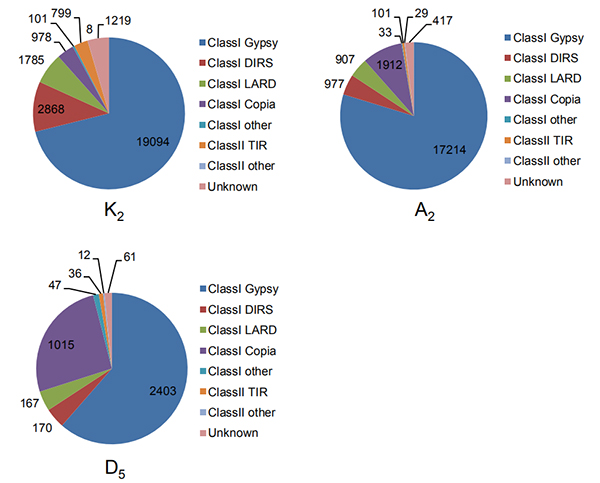
Wang M et al.,Molekularbiologie und Evolution, 2021
5.Hi-C-Wärmekarte desC. acuminataGenom, das genomweite Gesamtinteraktionen zeigt.Die Intensität der Hi-C-Wechselwirkungen ist proportional zum linearen Abstand zwischen Contigs.Eine saubere gerade Linie auf dieser Wärmekarte weist auf eine äußerst genaue Verankerung von Contigs auf Chromosomen hin.(Contig-Verankerungsverhältnis: 96,03 %)
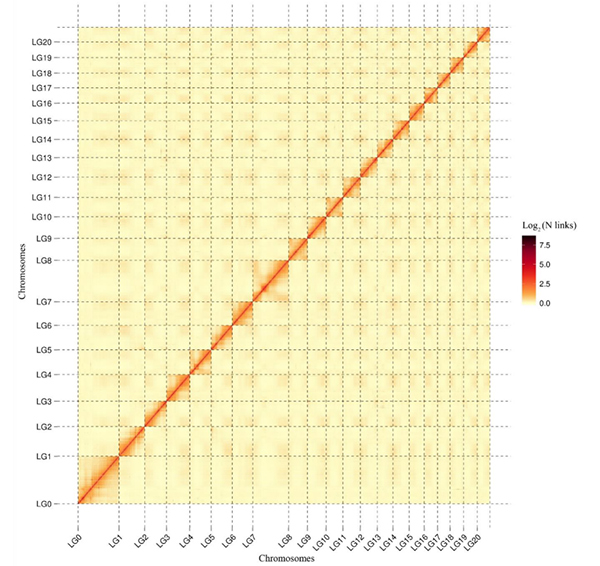
kang M et al.,Naturkommunikation,2021
BMK-Fall
Eine qualitativ hochwertige Genomassemblierung hebt die genomischen Merkmale und agronomisch wichtigen Gene des Roggens hervor
Veröffentlicht: Naturgenetik, 2021
Sequenzierungsstrategie:
Genomassemblierung: PacBio CLR-Modus mit 20-kb-Bibliothek (497 GB, ca. 63×)
Sequenzkorrektur: NGS mit 270 bp DNA-Bibliothek (430 Gb, ca. 54×) auf Illumina-Plattform
Contigs-Verankerung: Hi-C-Bibliothek (560 GB, ca. 71×) auf der Illumina-Plattform
Optische Karte: (779,55 GB, ca. 99×) auf Bionano Irys
Wichtigste Ergebnisse
1. Eine Zusammenstellung des Weining-Roggengenoms wurde mit einer Gesamtgenomgröße von 7,74 GB veröffentlicht (98,74 % der durch Durchflusszytometrie geschätzten Genomgröße).Scaffold N50 dieser Baugruppe erreichte 1,04 GB.93,67 % der Contigs wurden erfolgreich auf 7 Pseudochromosomen verankert.Diese Baugruppe wurde mittels Linkage Map, LAI und BUSCO bewertet, was in allen Bewertungen zu hohen Punktzahlen führte.
2. Weitere Studien zur vergleichenden Genomik, zur Karte der genetischen Verknüpfung und zur Transkriptomik wurden auf der Grundlage dieses Genoms durchgeführt.Es wurde eine Reihe genomischer Merkmale aufgedeckt, darunter genomweite Genduplikationen und deren Einfluss auf Stärkebiosynthesegene;physikalische Organisation komplexer Prolamin-Loci, Genexpressionsmerkmale, die dem frühen Kopfmerkmal zugrunde liegen, und mutmaßliche domestizierungsassoziierte chromosomale Regionen und Loci im Roggen.
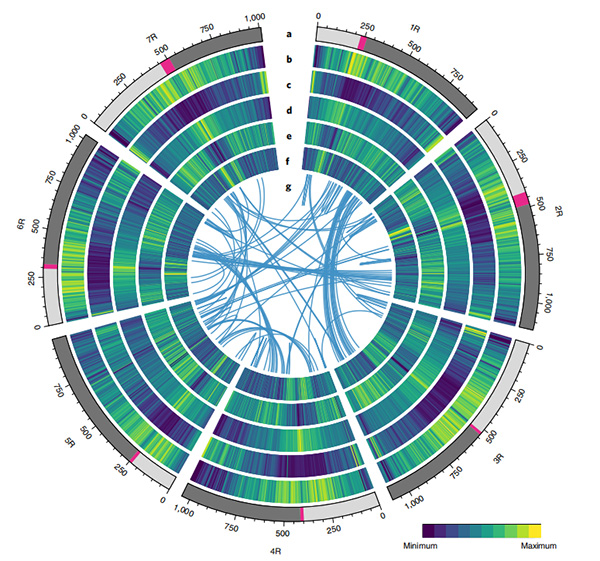 Circos-Diagramm zu genomischen Merkmalen des Weining-Roggengenoms | 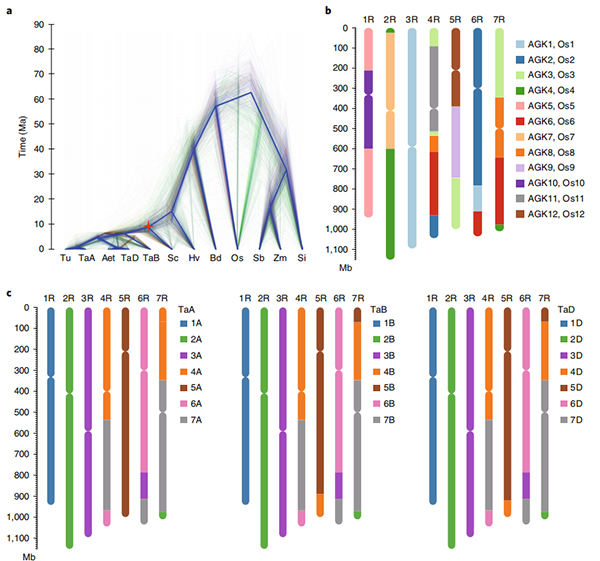 Evolutions- und Chromosomensyntenieanalysen des Roggengenoms |
Li, G., Wang, L., Yang, J.et al.Eine qualitativ hochwertige Genomassemblierung hebt die genomischen Merkmale und agronomisch wichtigen Gene des Roggens hervor.Nat Genet 53,574–584 (2021).
https://doi.org/10.1038/s41588-021-00808-z





