

Plant/Animal De Novo Genome Sequencing




Servicefördelar
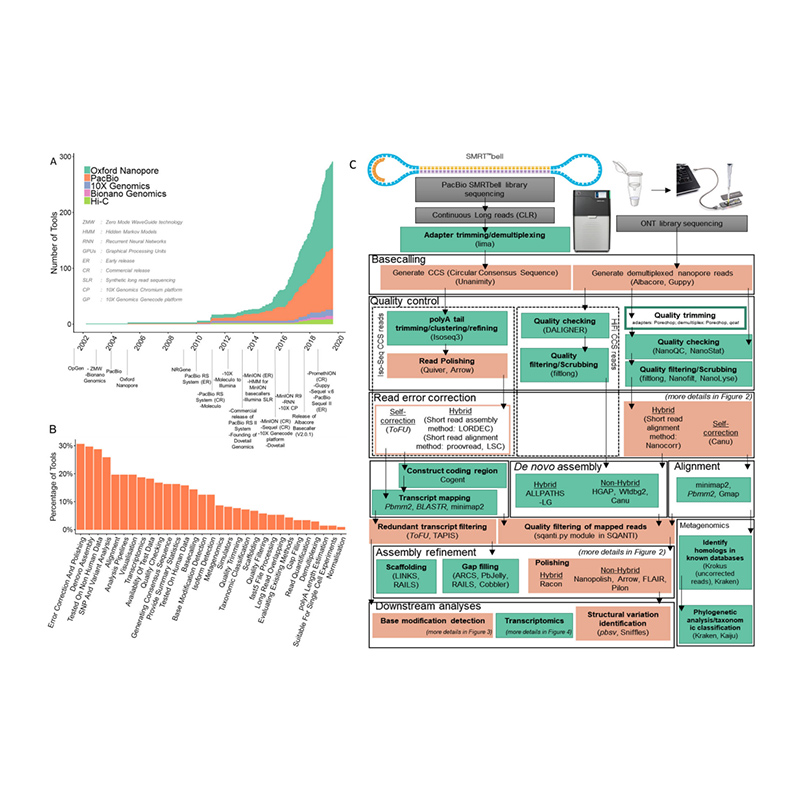
Utveckling av sekvenseringsplattformar och bioinformatik inomde novogenom sammansättning
(Amarasinghe SL et al.,Genombiologi, 2020)
● Konstruera nya genom och förbättra befintliga referensgenom för arter av intresse.
● Högre noggrannhet, kontinuitet och fullständighet vid montering
● Konstruera grundläggande resurs för forskning inom sekvenspolymorfism, QTLs, genredigering, förädling, etc.
● Utrustad med ett helt spektrum av tredje generationens sekvenseringsplattformar: en lösning för genommontering
● Flexibla sekvenserings- och sammansättningsstrategier som uppfyller olika genom med olika egenskaper
● Mycket skickligt bioinformatikerteam med stor erfarenhet av komplexa genomsammansättningar, inklusive polyploider, jättegenom etc.
● Över 100 framgångsrika fall med en ackumulerad publicerad effektfaktor på över 900
● Omläggningstid så snabbt som 3 månader för genomsamling på kromosomnivå.
● Gedigen teknisk support med en rad patent och mjukvaruupphovsrätter inom både experimentell sida och bioinformatik.
Servicespecifikationer
|
Innehåll
|
Plattform
|
Läs Längd
|
Rapportering
|
| Genomundersökning
| Illumina NovaSeq
| PE150
| ≥ 50X
|
| Genomsekvensering
| PacBio Revio
| 15 kb HiFi Läser
| ≥ 30X
|
| Hi-C
| Illumina NovaSeq
| PE150
| ≥100X
|
Arbetsflöde

Exempel på krav och leverans
Exempelkrav:
| Arter | Vävnad | För PacBio | För Nanopore |
| Djur | Viscerala organ (lever, mjälte, etc.) | ≥ 1,0 g | ≥ 3,5 g |
| Muskel | ≥ 1,5 g | ≥ 5,0 g | |
| Blod från däggdjur | ≥ 1,5 ml | ≥ 5,0 ml | |
| Blod från fiskar eller fåglar | ≥ 0,2 ml | ≥ 0,5 ml | |
| Växter | Färska blad | ≥ 1,5 g | ≥ 5,0 g |
| Kronblad eller stjälk | ≥ 3,5 g | ≥ 10,0 g | |
| Rötter eller frön | ≥ 7,0 g | ≥ 20,0 g | |
| Celler | Cell kultur | ≥ 3×107 | ≥ 1×108 |
Rekommenderad provleverans
Behållare: 2 ml centrifugrör (tunnfolie rekommenderas inte)
För de flesta prover rekommenderar vi att inte konservera i etanol.
Provmärkning: Proverna måste vara tydligt märkta och identiska med det inlämnade provinformationsformuläret.
Försändelse: Torris: Proverna måste först packas i påsar och grävas ner i torris.
Service arbetsflöde

Experimentdesign

Provleverans

DNA-extraktion

Byggande av bibliotek

Sekvensering

Dataanalys

Service efter försäljning
*Demoresultat som visas här är alla från genom publicerade med Biomarker Technologies
1.Circos på kromosom-nivå genom montering avG. rotundifoliumav Nanopore sekvenseringsplattform
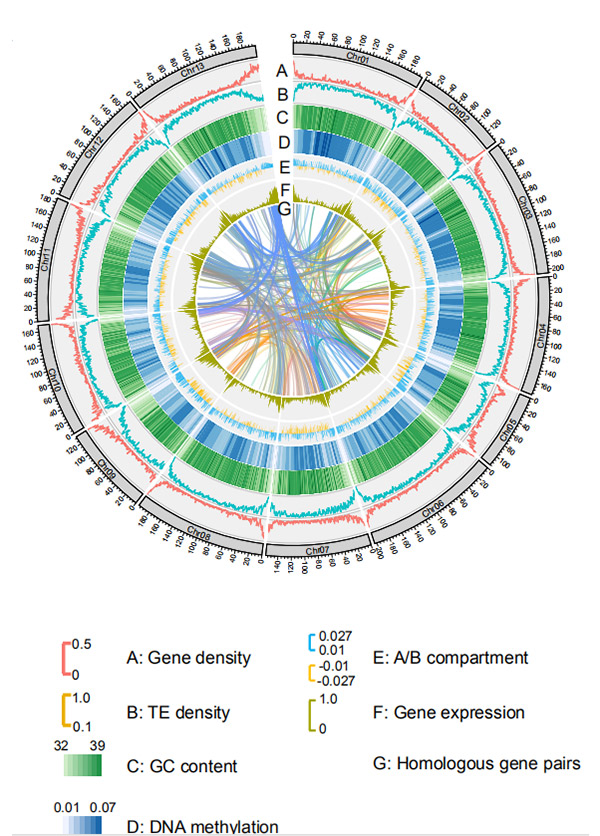
Wang M et al.,Molekylärbiologi och evolution, 2021
2. Statistik över Weining råg genom montering och anteckning
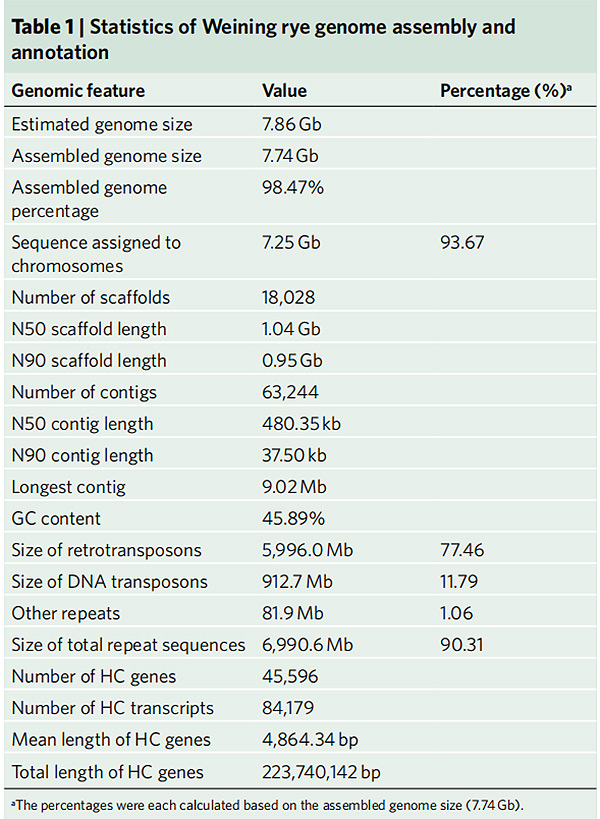
Li G et al.,Naturgenetik, 2021
3.Genförutsägelse avSechium edulegenomet, härlett från tre förutsägelsesmetoder:De novoförutsägelse, homologi-baserad förutsägelse och RNA-Seq-databaserad förutsägelse
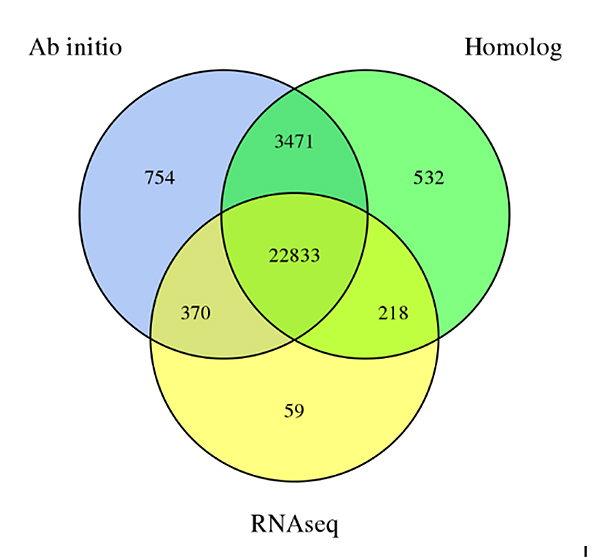
Fu A et al.,Trädgårdsbruksforskning, 2021
4.Identifiering av intakta långa terminala upprepningar i tre bomullsgenom
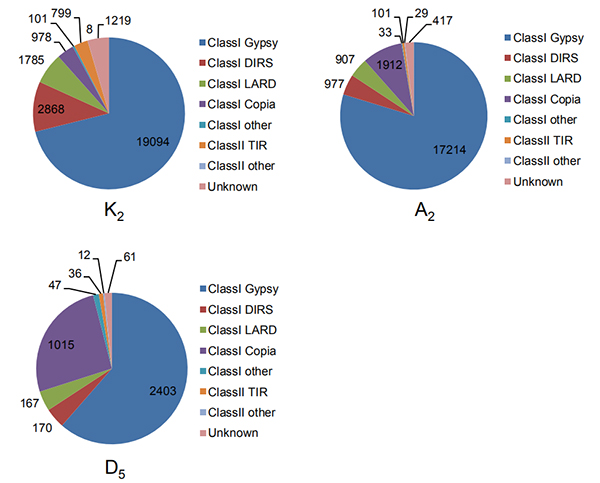
Wang M et al.,Molekylärbiologi och evolution, 2021
5.Hi-C värmekarta överC. acuminatagenom som visar genomgående interaktioner.Intensiteten hos Hi-C-interaktioner är proportionell mot linjärt avstånd mellan kontiger.En ren rak linje på denna värmekarta indikerar en mycket noggrann förankring av sammanhängningar på kromosomerna.(Contig-förankringsförhållande: 96,03 %)
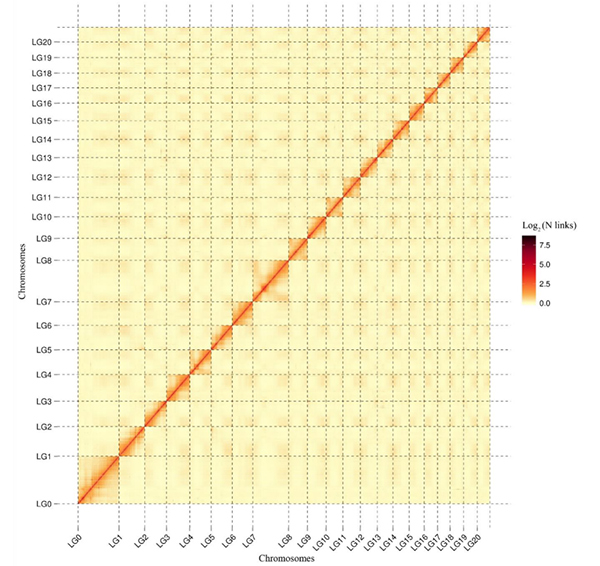
kang M et al.,Naturkommunikation,2021
BMK Fall
En genomsamling av hög kvalitet framhäver rågens genomiska egenskaper och agronomiskt viktiga gener
Publicerad: Naturgenetik, 2021
Sekvenseringsstrategi:
Genomsammansättning: PacBio CLR-läge med 20 kb bibliotek (497 Gb, ca 63×)
Sekvenskorrigering: NGS med 270 bp DNA-bibliotek (430 Gb, ca 54×) på Illumina-plattformen
Contigs förankring: Hi-C-bibliotek (560 Gb, ca 71×) på Illumina-plattformen
Optisk karta: (779,55 Gb, ca 99×) på Bionano Irys
Nyckelresultat
1. En samling av Weining råggenom publicerades med total genomstorlek på 7,74 Gb (98,74% av uppskattad genomstorlek genom flödescytometri).Ställningen N50 i denna enhet uppnådde 1,04 Gb.93,67% av contigs var framgångsrikt förankrade på 7 pseudokromosomer.Denna sammansättning utvärderades av länkkarta, LAI och BUSCO, vilket resulterade i höga poäng i alla utvärderingar.
2. Ytterligare studier på jämförande genomik, genetisk kopplingskarta, transkriptomiska studier utfördes på basen av detta genom.En serie egenskaper relaterade genomiska egenskaper avslöjades inklusive genomomfattande gendupliceringar och deras inverkan på stärkelsebiosyntesgener;fysisk organisation av komplexa prolaminloki, genuttrycksegenskaper bakom tidig rubrikegenskaper och förmodade domesticeringsassocierade kromosomala regioner och loci i råg.
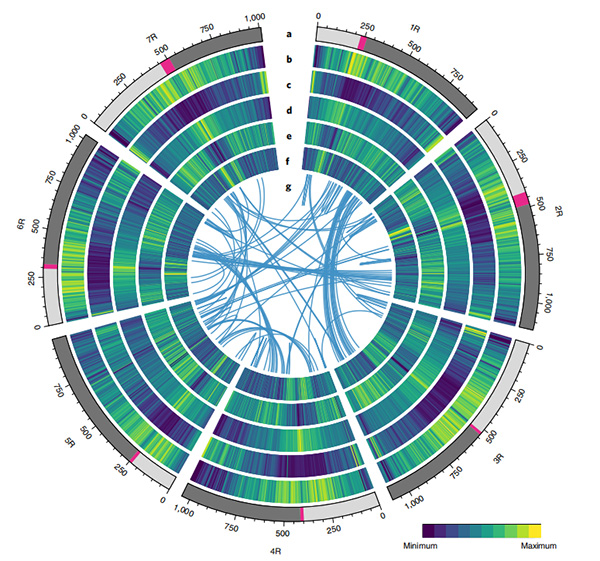 Circos diagram på genomiska egenskaper hos Weining råg genom | 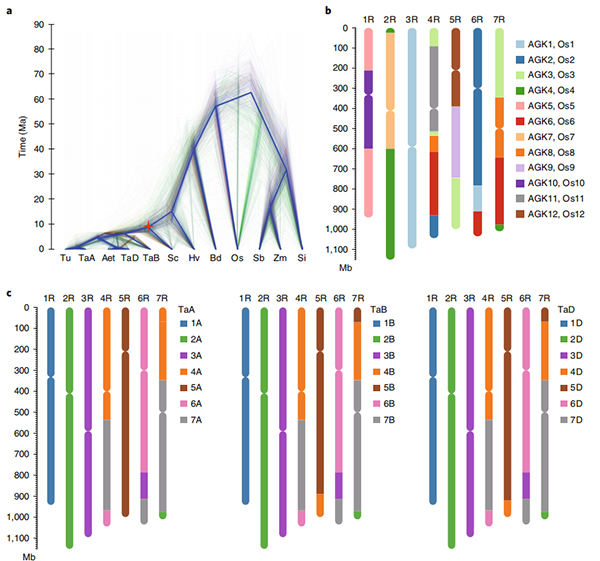 Evolutions- och kromosomsyntenyanalyser av rågens genom |
Li, G., Wang, L., Yang, J.et al.En genomsamling av hög kvalitet framhäver rågens genomiska egenskaper och agronomiskt viktiga gener.Nat Genet 53,574–584 (2021).
https://doi.org/10.1038/s41588-021-00808-z





