

Nicht referenzbasierte mRNA-Sequenzierung – Illumina








Merkmale
● Unabhängig von einem Referenzgenom,
● Die Daten könnten zur Analyse der Struktur und des Ausdrucks von Transkripten verwendet werden
● Identifizieren Sie variable Clipping-Sites
Servicevorteile
● BMKCloud-basierte Ergebnisbereitstellung: Die Ergebnisse werden als Datendatei und interaktiver Bericht über die BMKCloud-Plattform bereitgestellt, was ein benutzerfreundliches Lesen komplexer Analyseausgaben und individuelles Data Mining auf der Grundlage standardmäßiger Bioinformatikanalysen ermöglicht.
● Kundendienst: Kundendienst, gültig für 3 Monate nach Projektabschluss, einschließlich Projektnachverfolgung, Fehlerbehebung, Fragen und Antworten zu Ergebnissen usw.
Musteranforderungen und Lieferung
Probenanforderungen:
Nukleotide:
| Konz. (ng/μl) | Menge (μg) | Reinheit | Integrität |
| ≥ 20 | ≥ 0,5 | OD260/280=1,7-2,5 OD260/230=0,5-2,5 Begrenzte oder keine Protein- oder DNA-Kontamination auf dem Gel sichtbar. | Für Pflanzen: RIN≥6,5; Für Tiere: RIN≥7,0; 5,0≥28S/18S≥1,0; begrenzte oder keine Grundlinienhöhe |
Gewebe: Gewicht (trocken): ≥1 g
*Für Gewebe mit weniger als 5 mg empfehlen wir, schockgefrorene (in flüssigem Stickstoff) Gewebeproben einzusenden.
Zellsuspension: Zellzahl = 3×107
*Wir empfehlen den Versand von gefrorenem Zelllysat.Für den Fall, dass die Zellenzahl kleiner als 5×10 ist5Es wird empfohlen, in flüssigem Stickstoff schockgefrostet zu werden.
Blutproben:
PA×geneBloodRNATube;
6 mlTRIzol und 2 ml Blut (TRIzol:Blut=3:1)
Empfohlene Musterlieferung
Container:
2 ml Zentrifugenröhrchen (Alufolie wird nicht empfohlen)
Probenbeschriftung: Gruppe+Replikation, z. B. A1, A2, A3;B1, B2, B3... ...
Sendung:
1. Trockeneis: Proben müssen in Beutel verpackt und in Trockeneis vergraben werden.
2.RNAstable-Röhrchen: RNA-Proben können in RNA-Stabilisierungsröhrchen (z. B. RNAstable®) getrocknet und bei Raumtemperatur versendet werden.
Service-Workflow

Experimentdesign

Musterlieferung

RNA-Extraktion

Bibliotheksbau

Sequenzierung

Datenanalyse

Kundendienst
Bioinformatik
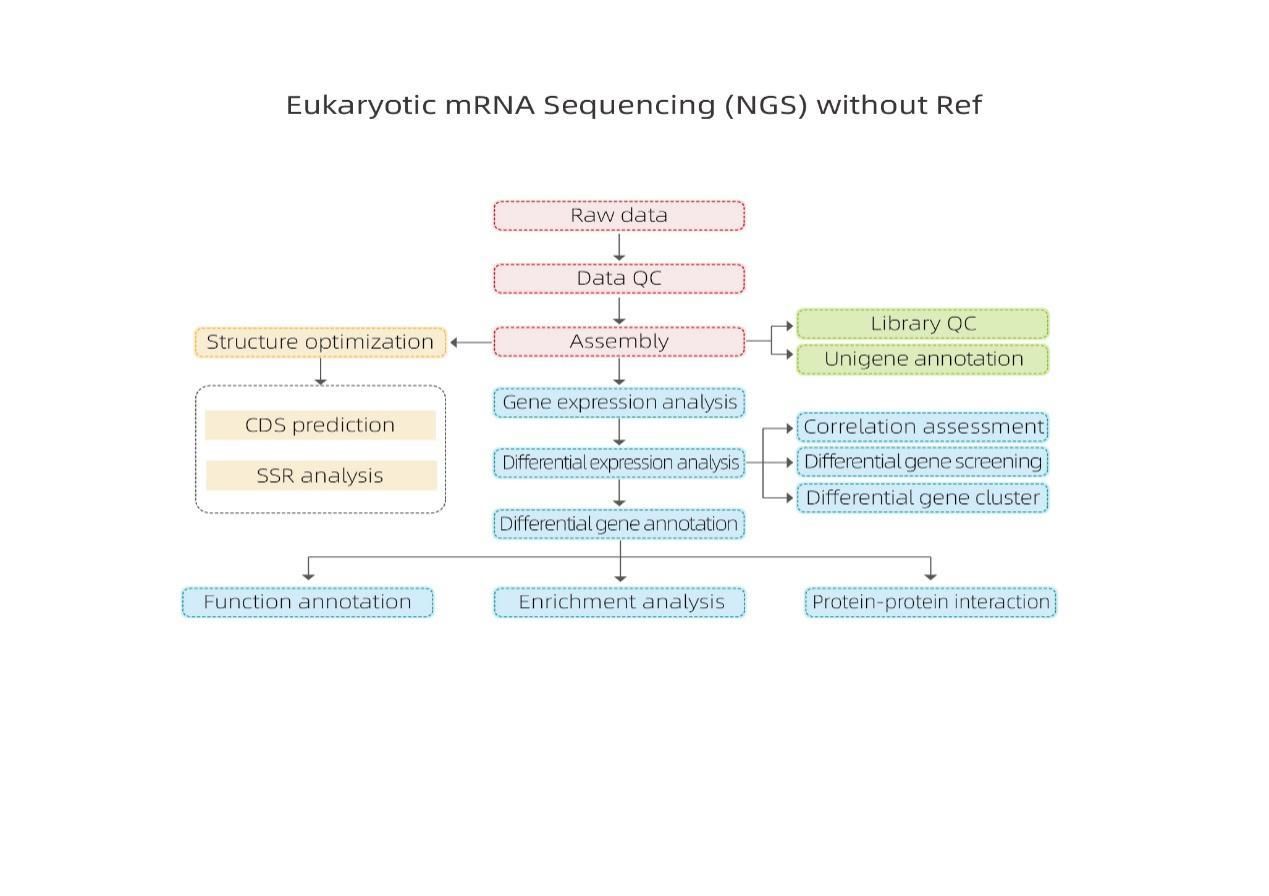
1.mRNA(denovo)-Assemblierungsprinzip
Bei Trinity werden Lesevorgänge in kleinere Teile fragmentiert, die als K-mer bekannt sind.Diese K-mers werden dann als Seeds verwendet, die in Contigs erweitert werden und dann basierend auf Contig-Überlappungen Komponenten bilden.Schließlich wurde hier De Bruijn angewendet, um Transkripte in den Komponenten zu erkennen.
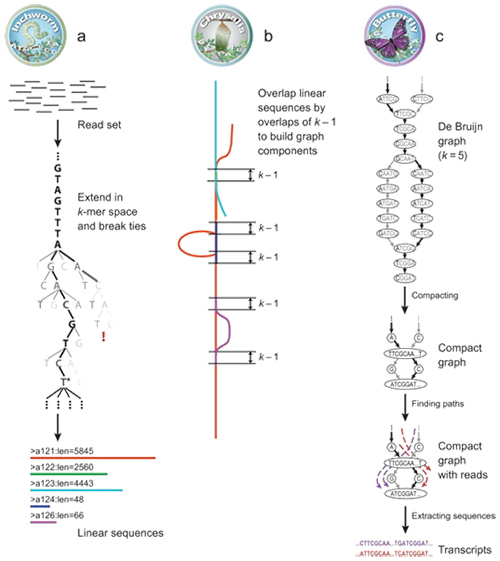
mRNA (De novo) Übersicht über Trinity
2. mRNA-Verteilung (De novo) des Genexpressionsniveaus
RNA-Seq ist in der Lage, eine hochempfindliche Schätzung der Genexpression zu erreichen.Normalerweise liegt der nachweisbare Bereich der Transkriptexpression FPKM im Bereich von 10^-2 bis 10^6
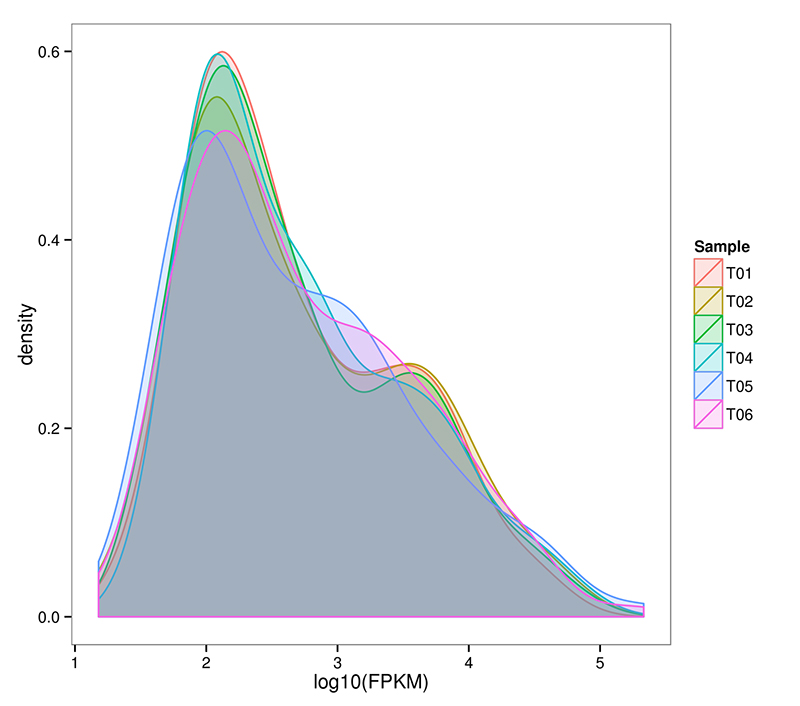
mRNA (De novo) Verteilung der FPKM-Dichte in jeder Probe
3.mRNA (De novo) GO-Anreicherungsanalyse von DEGs
Die GO-Datenbank (Gene Ontology) ist ein strukturiertes biologisches Annotationssystem, das ein Standardvokabular der Funktionen von Genen und Genprodukten enthält.Es enthält mehrere Ebenen, wobei die Funktionen umso spezifischer sind, je niedriger die Ebene ist.
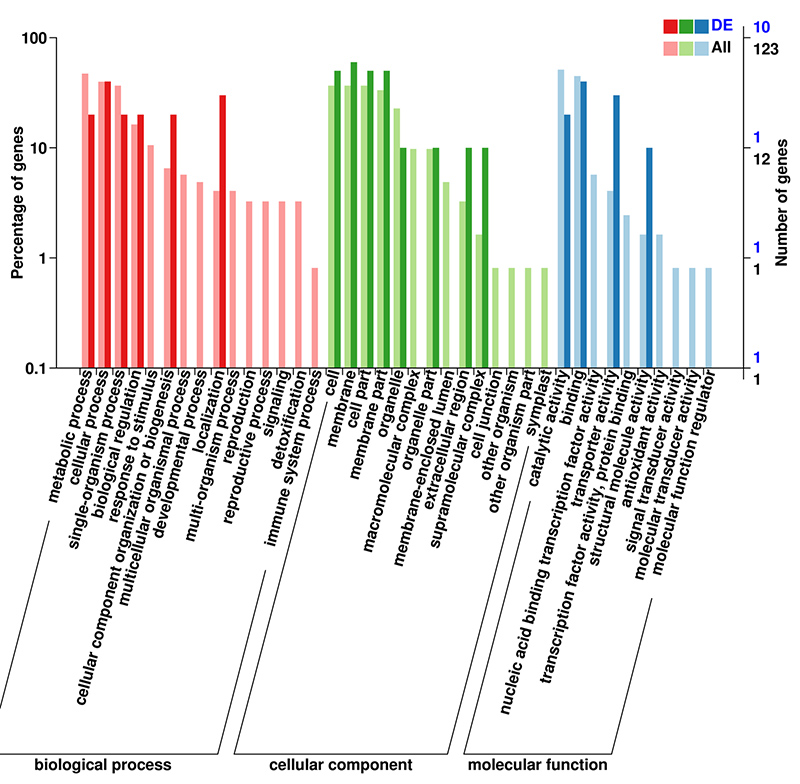
mRNA (De novo) GO-Klassifizierung von DEGs auf der zweiten Ebene
BMK-Fall
Transkriptomanalyse des Saccharosestoffwechsels während der Schwellung und Entwicklung der Zwiebeln bei Zwiebeln (Allium cepa L.)
Veröffentlicht: Grenzen der Pflanzenwissenschaft,2016
Sequenzierungsstrategie
Illumina HiSeq2500
Beispielsammlung
In dieser Studie wurde die Sorte „Utah Yellow Sweet Spain“ „Y1351“ verwendet.Die Anzahl der gesammelten Proben betrug
15. Tag nach Schwellung (DAS) der Knolle (2 cm Durchmesser und 3–4 g Gewicht), 30. DAS (5 cm Durchmesser und 100–110 g Gewicht) und ∼3 am 40. DAS (7 cm Durchmesser und 260–300 Gramm).
Wichtigste Ergebnisse
1. Im Venn-Diagramm wurden insgesamt 146 DEGs über alle drei Paare von Entwicklungsstadien hinweg nachgewiesen
2. „Kohlenhydrattransport und -stoffwechsel“ war nur durch 585 Unigene vertreten (dh 7 % des annotierten COG).
3.Unigenes, die erfolgreich in der GO-Datenbank annotiert wurden, wurden in drei Hauptkategorien für die drei verschiedenen Stadien der Zwiebelentwicklung eingeteilt.In der Hauptkategorie „biologischer Prozess“ waren „Stoffwechselprozesse“ am häufigsten vertreten, gefolgt von „zellulären Prozessen“.In der Hauptkategorie „molekulare Funktion“ waren die beiden Kategorien „Bindung“ und „katalytische Aktivität“ am häufigsten vertreten.
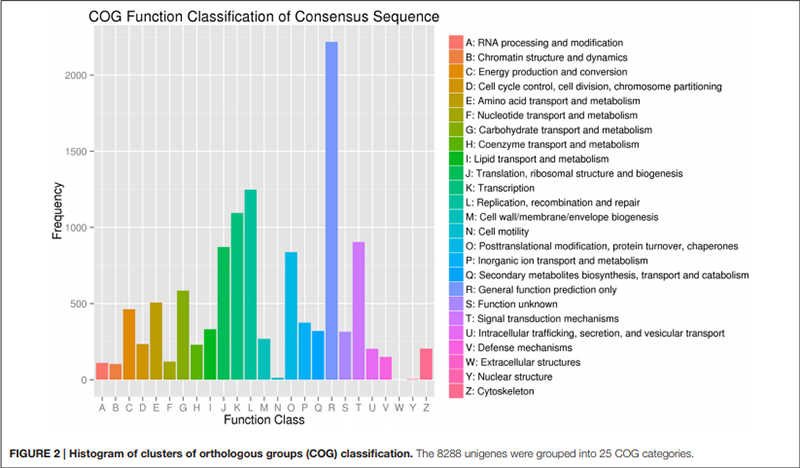 Histogramm der Klassifikation von Clustern orthologer Gruppen (COG). | 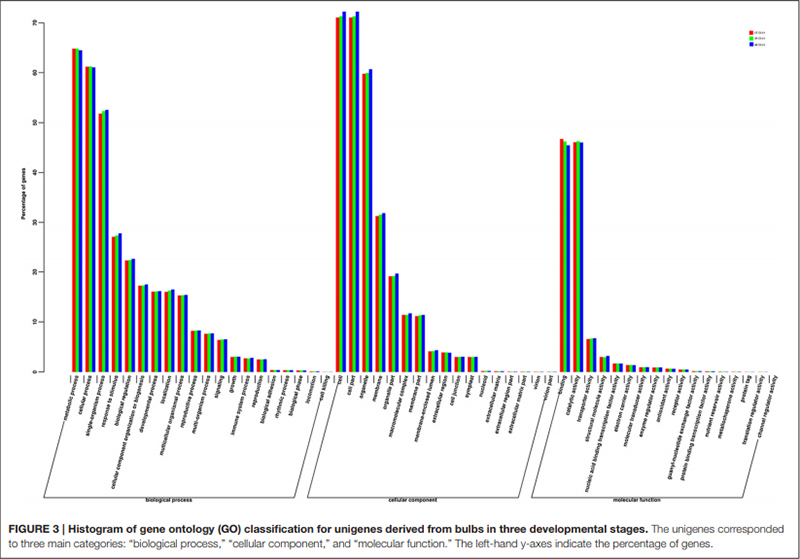 Histogramm der Klassifikation der Genontologie (GO) für Unigene, die aus Zwiebeln in drei Entwicklungsstadien stammen |
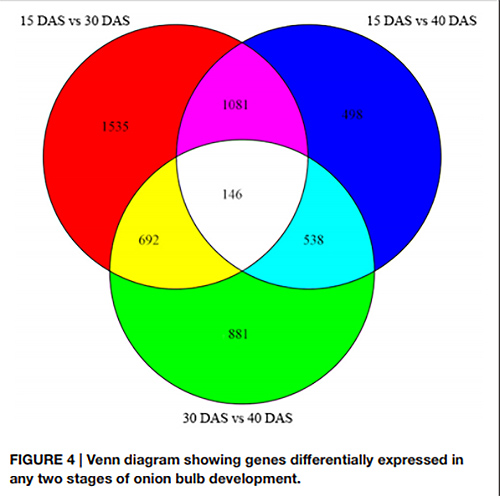 Venn-Diagramm, das Gene zeigt, die in zwei beliebigen Stadien der Zwiebelknollenentwicklung unterschiedlich exprimiert werden |
Referenz
Zhang C, Zhang H, Zhan Z, et al.Transkriptomanalyse des Saccharosestoffwechsels während der Schwellung und Entwicklung der Zwiebeln in Zwiebeln (Allium cepa L.)[J].Frontiers in Plant Science, 2016, 7:1425-.DOI: 10.3389/fpls.2016.01425





