

Sequenziamento di frammenti amplificati con locus specifico (SLAF-Seq)





Dettagli del servizio
Schema tecnico
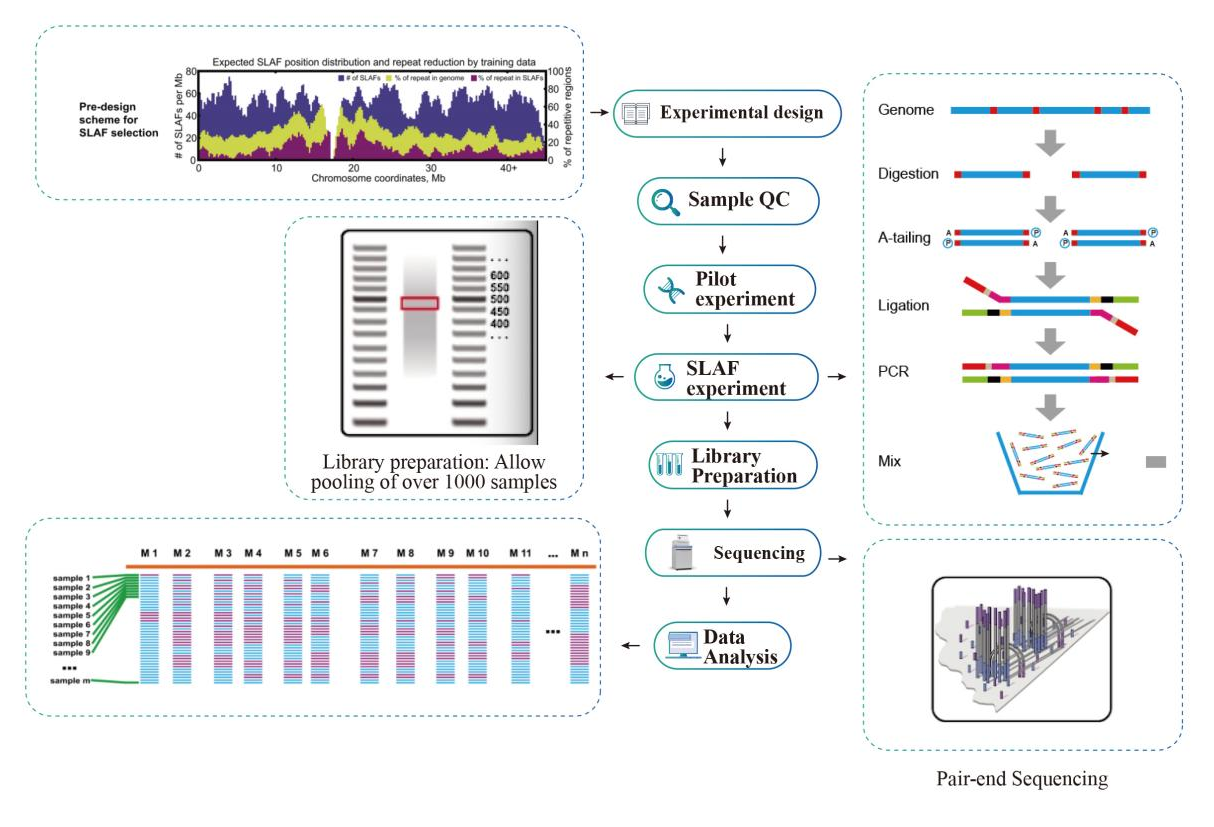
Flusso di lavoro

Vantaggi del servizio
Elevata efficienza di rilevamento dei marcatori- La tecnologia di sequenziamento ad alto rendimento aiuta SLAF-Seq a scoprire centinaia di migliaia di tag all'interno dell'intero genoma.
Bassa dipendenza dal genoma- Può essere applicato a specie con o senza genoma di riferimento.
Progettazione di schemi flessibili- Digestione a singolo enzima, doppio enzima, multi-enzima e vari tipi di enzimi, tutti possono essere selezionati per soddisfare diversi obiettivi di ricerca o specie.La pre-valutazione in silico viene utilizzata per garantire una progettazione enzimatica ottimale.
Digestione enzimatica efficiente- L'esperimento preliminare è stato effettuato per ottimizzare le condizioni, il che rende l'esperimento formale stabile e affidabile.L'efficienza della raccolta dei frammenti può raggiungere oltre il 95%.
Tag SLAF distribuiti uniformemente- I tag SLAF sono distribuiti uniformemente in tutti i cromosomi nella massima misura, raggiungendo una media di 1 SLAF per 4 kb.
Evitamento efficace delle ripetizioni- La sequenza ripetitiva nei dati SLAF-Seq è ridotta a meno del 5%, soprattutto nelle specie con un alto livello di ripetizioni, come grano, mais, ecc.
Vasta esperienza-Oltre 2000 progetti SLAF-Seq chiusi su centinaia di specie che comprendono piante, mammiferi, uccelli, insetti, organismi acquatici, ecc.
Flusso di lavoro bioinformatico autosviluppato- Un flusso di lavoro bioinformatico integrato per SLAF-Seq è stato sviluppato da BMKGENE per garantire l'affidabilità e l'accuratezza dell'output finale.
Specifiche del servizio
| piattaforma | Concentrato(ng/gl) | Totale (ug) | OD260/280 |
| Illumina NovaSeq | >35 | >1.6(Volume>15μl) | 1.6-2.5 |
Strategia di sequenziamento consigliata
Profondità di sequenziamento: 10X/Tag
| Dimensione del genoma | Tag SLAF consigliati |
| < 500 MB | 100K o WGS |
| 500 Mb- 1 Gb | 100K |
| 1 GB -2 GB | 200K |
| Genomi giganti o complessi | 300 - 400K |
| Applicazioni
| Consigliato Scala della popolazione
| Strategia e profondità del sequenziamento
| |
| Profondità
| Numero identificativo
| ||
| GWAS
| Numero di campioni ≥ 200
| 10X
|
Secondo dimensione del genoma
|
| Evoluzione genetica
| Individui di ciascuno sottogruppo ≥ 10; campioni totali ≥ 30
| 10X
| |
Consegna del campione consigliata
Contenitore: provetta da centrifuga da 2 ml
Per la maggior parte dei campioni si consiglia di non conservare in etanolo.
Etichettatura dei campioni: i campioni devono essere chiaramente etichettati e identici al modulo informativo del campione inviato.
Spedizione: Ghiaccio secco: i campioni devono essere prima imballati in sacchetti e sepolti nel ghiaccio secco.
Flusso di lavoro del servizio






Controllo qualità del campione
Esperimento pilota
Esperimento SLAF
Preparazione della biblioteca
Sequenziamento
Analisi dei dati
Servizi post-vendita
1. Statistiche del risultato della mappa

2. Sviluppo dei marcatori SLAF

3. Annotazione della variazione

| Anno | rivista | IF | Titolo | Applicazioni |
| 2022 | Comunicazioni della natura | 17.694 | Basi genomiche dei giga-cromosomi e giga-genoma della peonia arborea Peonia ostii | SLAF-GWAS |
| 2015 | Nuovo fitologo | 7.433 | Le impronte della domesticazione ancorano le regioni genomiche di importanza agronomica semi di soia | SLAF-GWAS |
| 2022 | Giornale di ricerca avanzata | 12.822 | Introgressioni artificiali su tutto il genoma di Gossypium barbadense in G. hirsutum rivelano loci superiori per il miglioramento simultaneo della qualità e della resa della fibra di cotone tratti | SLAF-Genetica evolutiva |
| 2019 | Pianta molecolare | 10.81 | L'analisi genomica della popolazione e l'assemblaggio de novo rivelano l'origine di Weedy Il riso come gioco evolutivo | SLAF-Genetica evolutiva |
| 2019 | Genetica della natura | 31.616 | Sequenza del genoma e diversità genetica della carpa comune, Cyprinus carpio | Mappa del collegamento SLAF |
| 2014 | Genetica della natura | 25.455 | Il genoma dell'arachide coltivata fornisce informazioni sui cariotipi dei legumi, poliploidi Evoluzione e domesticazione delle colture. | Mappa del collegamento SLAF |
| 2022 | Giornale delle biotecnologie vegetali | 9.803 | L'identificazione di ST1 rivela una selezione che coinvolge l'autostop della morfologia del seme e il contenuto di olio durante la domesticazione della soia | Sviluppo di marcatori SLAF |
| 2022 | Giornale internazionale di scienze molecolari | 6.208 | Identificazione e sviluppo di marcatori di DNA per un Wheat-Leymus mollis 2Ns (2D) Sostituzione cromosomica disomica | Sviluppo di marcatori SLAF |





