

Secuenciación de fragmentos amplificados de locus específico (SLAF-Seq)





Detalles del servicio
Esquema Técnico
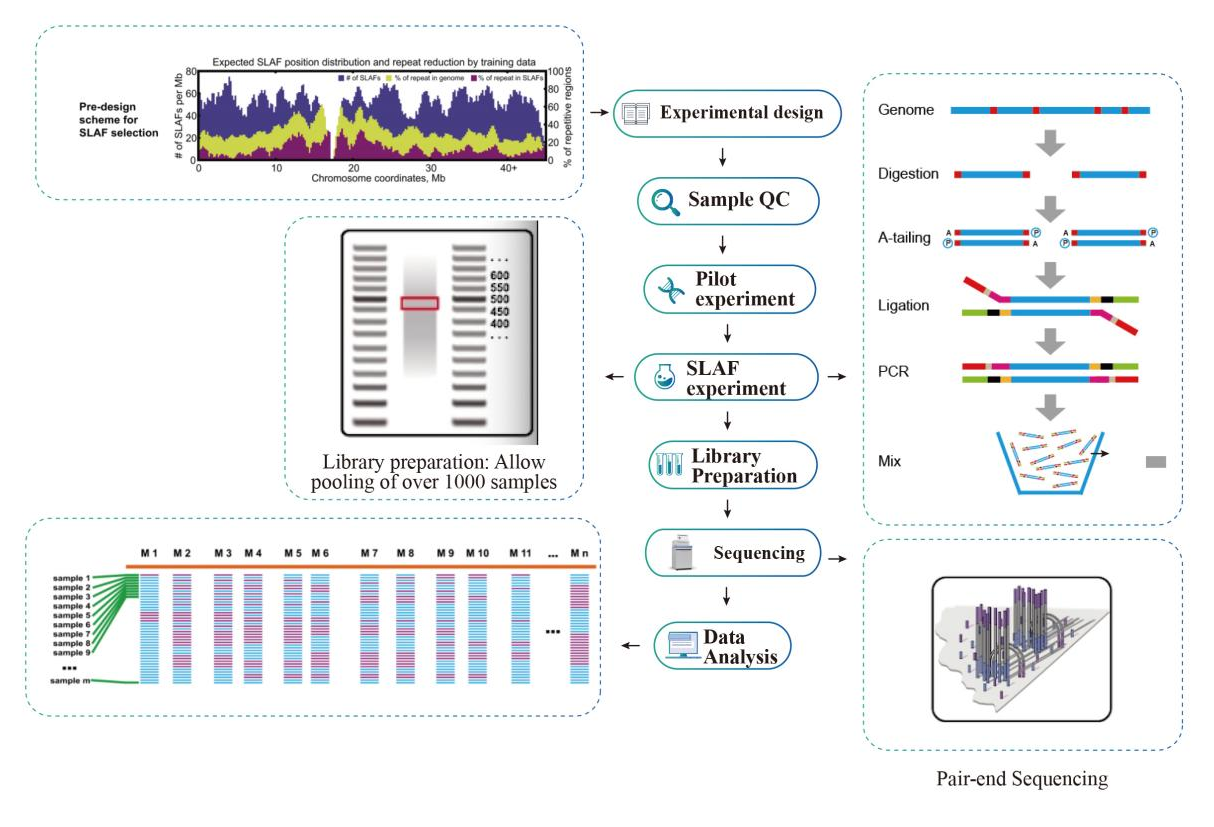
flujo de trabajo

Ventajas del servicio
Alta eficiencia de descubrimiento de marcadores- La tecnología de secuenciación de alto rendimiento ayuda a SLAF-Seq a descubrir cientos de miles de etiquetas dentro de todo el genoma.
Baja dependencia del genoma.- Puede aplicarse a especies con o sin genoma de referencia.
Diseño de esquema flexible- Digestión con una sola enzima, doble enzima, múltiples enzimas y varios tipos de enzimas, todas pueden seleccionarse para atender diferentes objetivos o especies de investigación.La evaluación previa in silico se utiliza para asegurar un diseño enzimático óptimo.
Digestión enzimática eficiente- Se llevó a cabo un experimento previo para optimizar las condiciones, lo que hace que el experimento formal sea estable y confiable.La eficiencia de la recolección de fragmentos puede alcanzar más del 95%.
Etiquetas SLAF distribuidas uniformemente- Las etiquetas SLAF se distribuyen uniformemente en todos los cromosomas en la mayor medida posible, logrando un promedio de 1 SLAF por 4 kb.
Evitación efectiva de repeticiones.- La secuencia repetitiva en los datos SLAF-Seq se reduce a menos del 5%, especialmente en especies con un alto nivel de repeticiones, como trigo, maíz, etc.
Amplia experiencia-Más de 2000 proyectos SLAF-Seq cerrados sobre cientos de especies que abarcan plantas, mamíferos, aves, insectos, organismos acuáticos, etc.
Flujo de trabajo bioinformático de desarrollo propio- BMKGENE desarrolló un flujo de trabajo bioinformático integrado para SLAF-Seq para garantizar la confiabilidad y precisión del resultado final.
Especificaciones de servicio
| Plataforma | Conc.(ng/gl) | Total (ug) | DE260/280 |
| Illumina NovaSeq | >35 | >1.6(Volumen>15μl) | 1,6-2,5 |
Estrategia de secuenciación recomendada
Profundidad de secuenciación: 10X/etiqueta
| Tamaño del genoma | Etiquetas SLAF recomendadas |
| < 500 MB | 100K o WGS |
| 500 Mb - 1 Gb | 100K |
| 1GB -2GB | 200K |
| Genomas gigantes o complejos | 300 - 400K |
| Aplicaciones
| Recomendado Escala de población
| Estrategia de secuenciación y profundidad.
| |
| Profundidad
| Número de etiqueta
| ||
| GWAS
| Número de muestra ≥ 200
| 10X
|
De acuerdo a tamaño del genoma
|
| Evolución genética
| individuos de cada subgrupo ≥ 10; muestras totales ≥ 30
| 10X
| |
Entrega de muestra recomendada
Envase: tubo de centrífuga de 2 ml
Para la mayoría de las muestras, recomendamos no conservarlas en etanol.
Etiquetado de muestras: las muestras deben estar claramente etiquetadas y ser idénticas al formulario de información de muestra enviado.
Envío: Hielo seco: las muestras deben empaquetarse primero en bolsas y enterrarse en hielo seco.
Flujo de trabajo del servicio






Control de calidad de muestra
experimento piloto
Experimento SLAF
Preparación de la biblioteca
Secuenciación
Análisis de los datos
Servicios postventa
1. Estadísticas del resultado del mapa.

2. Desarrollo del marcador SLAF

3. Anotación de variación

| Año | Diario | IF | Título | Aplicaciones |
| 2022 | Comunicaciones de la naturaleza | 17.694 | Base genómica de los gigacromosomas y el gigagenoma de la peonía arbórea. Paeonia ostii | SLAF-GWAS |
| 2015 | Nuevo fitólogo | 7.433 | Las huellas de domesticación anclan regiones genómicas de importancia agronómica en soja | SLAF-GWAS |
| 2022 | Revista de investigación avanzada | 12.822 | Introgresiones artificiales de todo el genoma de Gossypium barbadense en G. hirsutum revelan loci superiores para la mejora simultánea de la calidad y el rendimiento de la fibra de algodón rasgos | SLAF-Genética evolutiva |
| 2019 | Planta molecular | 10.81 | El análisis genómico de la población y el ensamblaje de novo revelan el origen de la maleza El arroz como juego evolutivo | SLAF-Genética evolutiva |
| 2019 | Genética de la naturaleza | 31.616 | Secuencia del genoma y diversidad genética de la carpa común, Cyprinus carpio. | Mapa de vinculación SLAF |
| 2014 | Genética de la naturaleza | 25.455 | El genoma del maní cultivado proporciona información sobre los cariotipos poliploides de las leguminosas Evolución y domesticación de cultivos. | Mapa de vinculación SLAF |
| 2022 | Revista de biotecnología vegetal | 9.803 | La identificación de ST1 revela una selección que implica hacer autostop en la morfología de las semillas y contenido de aceite durante la domesticación de la soja | Desarrollo de marcadores SLAF |
| 2022 | Revista Internacional de Ciencias Moleculares | 6.208 | Identificación y desarrollo de marcadores de ADN para un trigo-Leymus mollis 2Ns (2D) Sustitución de cromosomas disómicos | Desarrollo de marcadores SLAF |





