

ສະພາແຫ່ງ Genome ທີ່ອີງໃສ່ Hi-C

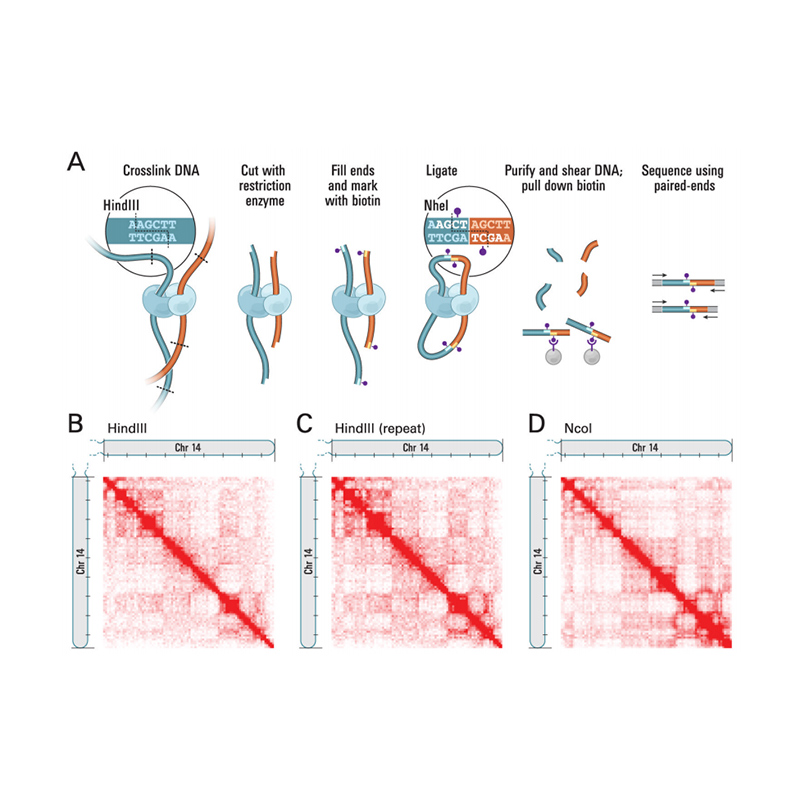






ຂໍ້ໄດ້ປຽບການບໍລິການ
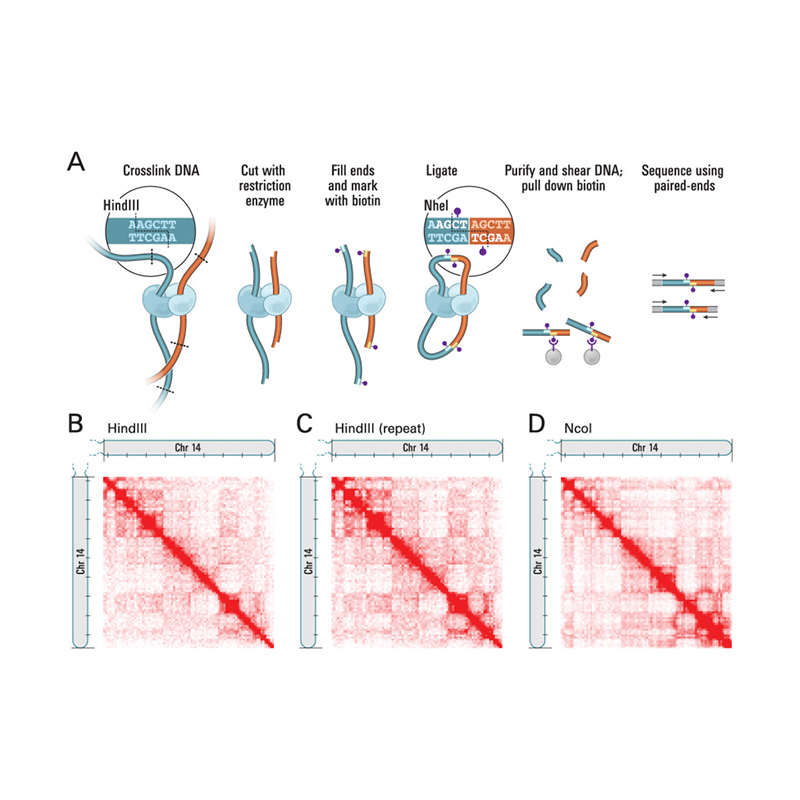
ພາບລວມຂອງ Hi-C
(Lieberman-Aiden E et al.,ວິທະຍາສາດ, 2009)
● ບໍ່ມີຄວາມຈໍາເປັນໃນການກໍ່ສ້າງປະຊາກອນພັນທຸກໍາສໍາລັບ contig anchoring;
● ຄວາມຫນາແຫນ້ນຂອງເຄື່ອງຫມາຍທີ່ສູງຂຶ້ນເຮັດໃຫ້ອັດຕາສ່ວນການຍຶດຕິດຂອງ contigs ສູງກວ່າ 90%;
● ເປີດໃຊ້ການປະເມີນຜົນ ແລະການແກ້ໄຂກ່ຽວກັບອົງປະກອບ genome ທີ່ມີຢູ່ແລ້ວ;
● ເວລາໝູນວຽນສັ້ນກວ່າ ພ້ອມກັບຄວາມຖືກຕ້ອງສູງໃນການປະກອບ genome;
● ປະສົບການທີ່ອຸດົມສົມບູນດ້ວຍຫ້ອງສະໝຸດ Hi-C ຫຼາຍກວ່າ 1000 ແຫ່ງທີ່ສ້າງຂຶ້ນຫຼາຍກວ່າ 500 ຊະນິດ;
● ຫຼາຍກວ່າ 100 ກໍລະນີທີ່ສໍາເລັດຜົນທີ່ມີປັດໄຈຜົນກະທົບການຈັດພີມມາສະສົມຂອງຫຼາຍກວ່າ 760;
● ການປະກອບ genome ທີ່ອີງໃສ່ Hi-C ສໍາລັບ genome polyploid, ອັດຕາສະມໍ 100% ແມ່ນບັນລຸໄດ້ໃນໂຄງການທີ່ຜ່ານມາ;
● ສິດທິບັດພາຍໃນ ແລະລິຂະສິດຊອບແວສໍາລັບການທົດລອງ Hi-C ແລະການວິເຄາະຂໍ້ມູນ;
● ຊອບແວການປັບຂໍ້ມູນແບບເປັນພາບທີ່ພັດທະນາດ້ວຍຕົວຕົນ, ຊ່ວຍໃຫ້ການເຄື່ອນຍ້າຍບລັອກດ້ວຍຕົນເອງ, ການຖອນຄືນ, ຖອນຄືນ ແລະເຮັດຄືນໃໝ່.
ຂໍ້ມູນຈໍາເພາະການບໍລິການ
|
ປະເພດຫ້ອງສະໝຸດ
|
ເວທີ | ອ່ານຄວາມຍາວ | ແນະນໍາຍຸດທະສາດ |
| Hi-C | Illumina NovaSeq | PE150 | ≥ 100X |
ການວິເຄາະ bioinformatics
● ການຄວບຄຸມຄຸນນະພາບຂໍ້ມູນດິບ
● ການຄວບຄຸມຄຸນນະພາບຫ້ອງສະໝຸດ Hi-C
● ການປະກອບ genome ທີ່ອີງໃສ່ Hi-C
● ການປະເມີນຜົນຫຼັງການປະກອບ
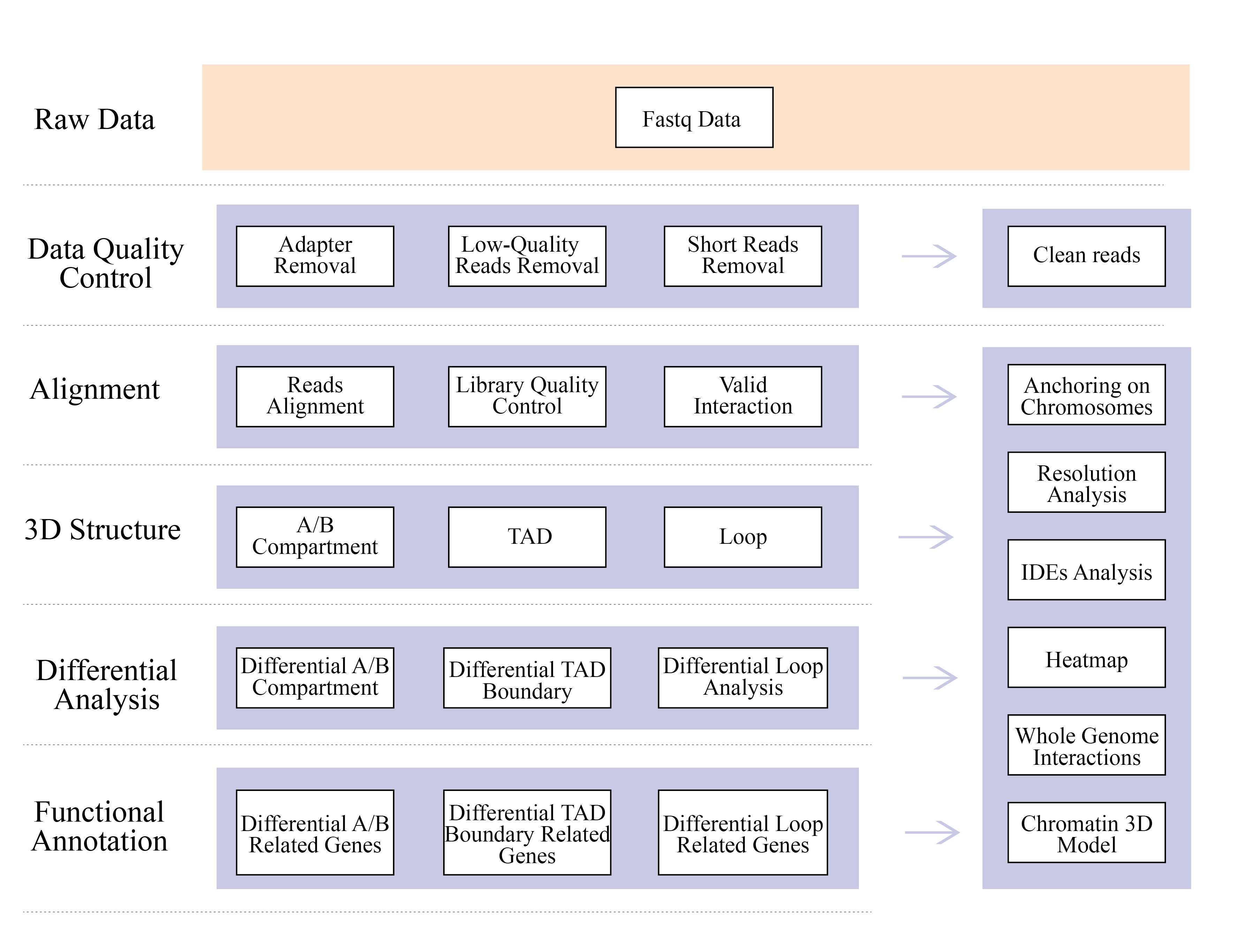
ຄວາມຕ້ອງການຕົວຢ່າງແລະການຈັດສົ່ງ
ຄວາມຕ້ອງການຕົວຢ່າງ:
| ສັດ | ເຊື້ອເຫັດ | ພືດ
|
| ເນື້ອເຍື່ອແຊ່ແຂງ: 1-2g ຕໍ່ຫ້ອງສະຫມຸດ ເຊລ: 1x 10^7 ເຊລຕໍ່ຫ້ອງສະໝຸດ | ເນື້ອເຍື່ອແຊ່ແຂງ: 1g ຕໍ່ຫ້ອງສະຫມຸດ | ເນື້ອເຍື່ອແຊ່ແຂງ: 1-2g ຕໍ່ຫ້ອງສະຫມຸດ
|
| *ພວກເຮົາແນະນຳໃຫ້ສົ່ງຢ່າງໜ້ອຍ 2 aliquots (1 g ແຕ່ລະອັນ) ສໍາລັບການທົດລອງ Hi-C. | ||
ການຈັດສົ່ງຕົວຢ່າງທີ່ແນະນໍາ
ບັນຈຸ: ທໍ່ centrifuge 2 ມລ (ບໍ່ແນະນໍາໃຫ້ໃຊ້ຟອຍກົ່ວ)
ສໍາລັບຕົວຢ່າງສ່ວນໃຫຍ່, ພວກເຮົາແນະນໍາໃຫ້ບໍ່ເກັບຮັກສາໃນເອທານອນ.
ການຕິດສະຫຼາກຕົວຢ່າງ: ຕົວຢ່າງຕ້ອງມີການຕິດສະຫຼາກຢ່າງຊັດເຈນ ແລະຄືກັນກັບແບບຟອມຂໍ້ມູນຕົວຢ່າງທີ່ສົ່ງມາ.
ການຂົນສົ່ງ: ນໍ້າກ້ອນແຫ້ງ: ຕົວຢ່າງຕ້ອງຖືກບັນຈຸໃສ່ຖົງກ່ອນ ແລະຝັງໄວ້ໃນນໍ້າກ້ອນແຫ້ງ.
ກະແສວຽກບໍລິການ

ການອອກແບບທົດລອງ

ການຈັດສົ່ງຕົວຢ່າງ

ການສະກັດເອົາ DNA

ການກໍ່ສ້າງຫໍສະຫມຸດ

ການຈັດລໍາດັບ

ການວິເຄາະຂໍ້ມູນ

ບໍລິການຫຼັງການຂາຍ
*ຜົນການສາທິດທີ່ສະແດງຢູ່ນີ້ແມ່ນມາຈາກ genomes ທີ່ເຜີຍແຜ່ດ້ວຍ Biomarker Technologies
ແຜນທີ່ຄວາມຮ້ອນປະຕິສໍາພັນ 1.Hi-C ຂອງCamptotheca acuminatagenome.ດັ່ງທີ່ສະແດງຢູ່ໃນແຜນທີ່, ຄວາມເຂັ້ມຂອງປະຕິສໍາພັນມີຄວາມສໍາພັນທາງລົບກັບໄລຍະທາງເສັ້ນ, ເຊິ່ງຊີ້ໃຫ້ເຫັນເຖິງການປະກອບລະດັບໂຄໂມໂຊມທີ່ມີຄວາມຖືກຕ້ອງສູງ.(ອັດຕາສ່ວນການຍຶດ: 96.03%)
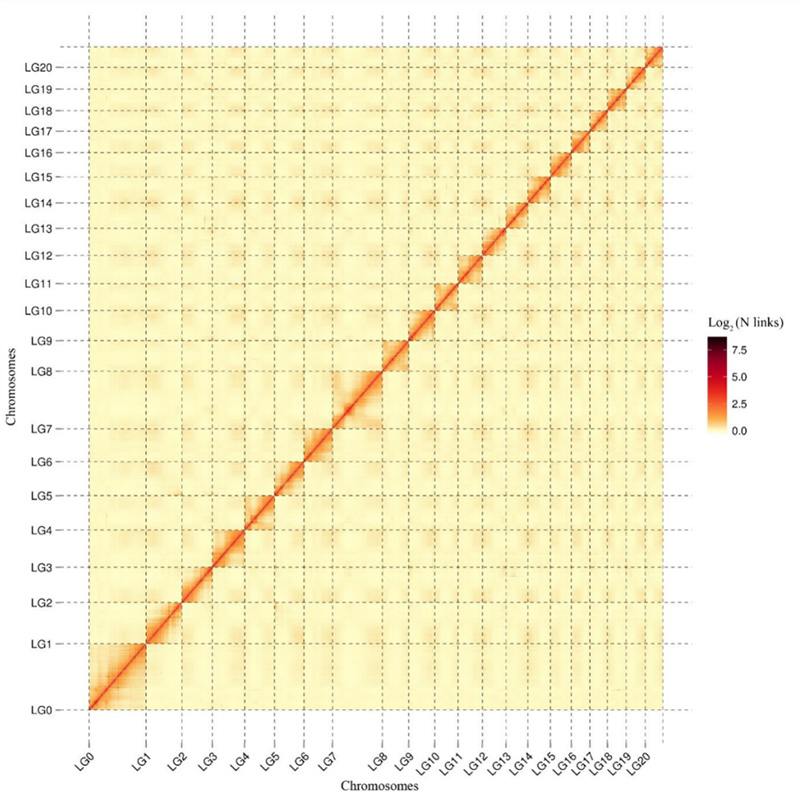
Kang M et al.,ການສື່ສານທໍາມະຊາດ, 2021
2.Hi-C ອໍານວຍຄວາມສະດວກໃນການກວດສອບຄວາມຖືກຕ້ອງຂອງ inversions ລະຫວ່າງGossypium hirsutumL. TM-1 A06 ແລະG. ສວນສັດChr06
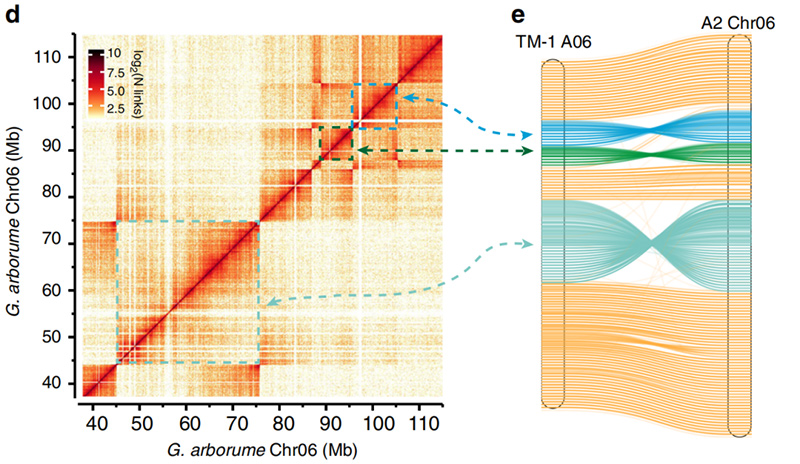
Yang Z et al.,ການສື່ສານທຳມະຊາດ, 2019
3.ການປະກອບ ແລະຄວາມແຕກຕ່າງ biallelic ຂອງ genome ມັນຕົ້ນ SC205.ແຜນທີ່ຄວາມຮ້ອນ Hi-C ສະແດງໃຫ້ເຫັນການແບ່ງປັນທີ່ຊັດເຈນຢູ່ໃນໂຄໂມໂຊມ homologous.
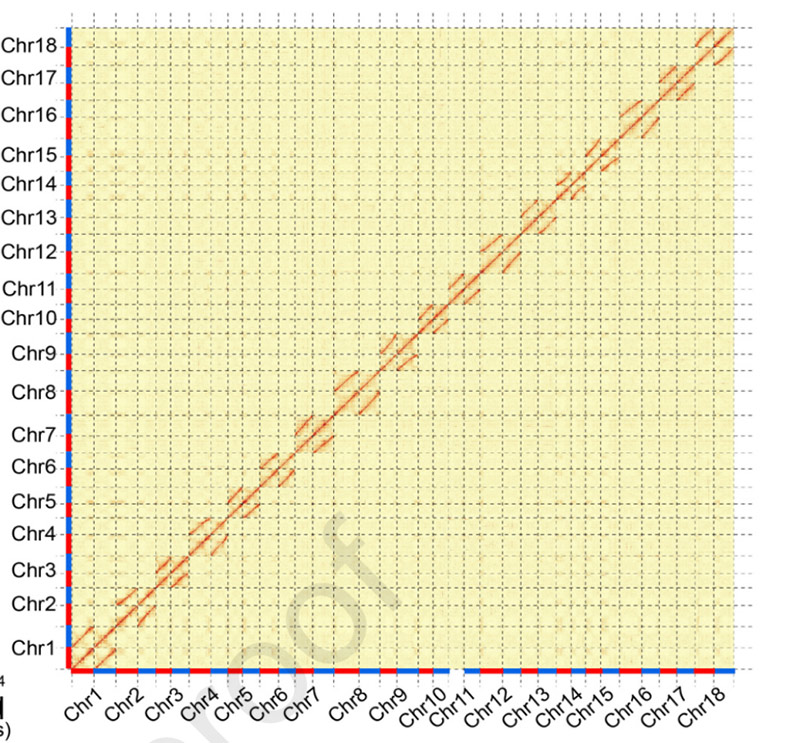
Hu W et al.,ພືດໂມເລກຸນ, 2021
ແຜນທີ່ຄວາມຮ້ອນ 4.Hi-C ກ່ຽວກັບການປະກອບ genome Ficus ສອງຊະນິດ:F.microcarpa(ອັດຕາສ່ວນ anchoring: 99.3%) ແລະF.hispida (ອັດຕາສ່ວນການຍຶດ: 99.7%)
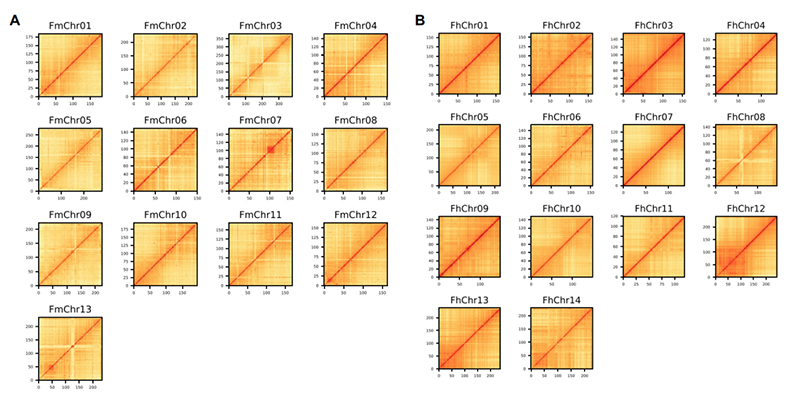
Zhang X et al.,ເຊລ, 2020
ກໍລະນີ BMK
ພັນທຸ ກຳ ຂອງຕົ້ນໄມ້ Banyan ແລະຕົວ pollinator Wasp ສະຫນອງຄວາມເຂົ້າໃຈກ່ຽວກັບ Coevolution Fig-wasp
ຈັດພີມມາ: ເຊລ, 2020
ຍຸດທະສາດການຈັດລໍາດັບ:
F. microcarpa genome: ປະມານ.84 X PacBio RSII (36.87 Gb) + Hi-C (44 Gb)
F. hispidagenome: ປະມານ.97 X PacBio RSII (36.12 Gb) + Hi-C (60 Gb)
Eupristina verticillatagenome: ປະມານ.170 X PacBio RSII (65 Gb)
ຜົນໄດ້ຮັບທີ່ສໍາຄັນ
1. ສອງພັນທຸກໍາຂອງຕົ້ນໄມ້ຕົ້ນກ້ວຍ ແລະ genome wasp pollinator ຫນຶ່ງໄດ້ຖືກກໍ່ສ້າງໂດຍໃຊ້ PacBio sequencing, Hi-C ແລະແຜນທີ່ການເຊື່ອມໂຍງ.
(1)F. microcarpagenome: ການປະກອບຂອງ 426 Mb (97.7% ຂອງຂະຫນາດ genome ຄາດຄະເນ) ໄດ້ຖືກສ້າງຕັ້ງຂຶ້ນດ້ວຍ contig N50 ຂອງ 908 Kb, ຄະແນນ BUSCO ຂອງ 95.6%.ໃນຈໍານວນທັງຫມົດ 423 Mb ລໍາດັບໄດ້ຖືກຍຶດຕິດກັບ 13 ໂຄໂມໂຊມໂດຍ Hi-C.ຄໍາອະທິບາຍກ່ຽວກັບພັນທຸກໍາໃຫ້ 29,416 genes-coding ທາດໂປຼຕີນ.
(2)F. Hispidagenome: ການປະກອບຂອງ 360 Mb (97.3% ຂອງຂະຫນາດ genome ຄາດຄະເນ) ແມ່ນຜົນຜະລິດທີ່ມີ contig N50 ຂອງ 492 Kb ແລະຄະແນນ BUSCO ຂອງ 97.4%.ລຳດັບທັງໝົດ 359 Mb ໄດ້ຖືກຍຶດໄວ້ໃນໂຄໂມໂຊມ 14 ໂຄໂມໂຊມໂດຍ Hi-C ແລະຄ້າຍຄືກັນກັບແຜນທີ່ການເຊື່ອມໂຍງທີ່ມີຄວາມໜາແໜ້ນສູງ.
(3)Eupristina verticillatagenome: ການປະກອບຂອງ 387 Mb (ຂະຫນາດ genome ຄາດຄະເນ: 382 Mb) ຖືກສ້າງຕັ້ງຂຶ້ນດ້ວຍ contig N50 ຂອງ 3.1 Mb ແລະຄະແນນ BUSCO ຂອງ 97.7%.
2.ການວິເຄາະ genomics ປຽບທຽບໄດ້ເປີດເຜີຍຈໍານວນທີ່ຍິ່ງໃຫຍ່ຂອງການປ່ຽນແປງໂຄງປະກອບການລະຫວ່າງສອງFicusgenomes, ເຊິ່ງສະຫນອງຊັບພະຍາກອນພັນທຸກໍາທີ່ບໍ່ມີຄ່າສໍາລັບການສຶກສາວິວັດທະນາການປັບຕົວ.ການສຶກສານີ້, ເປັນຄັ້ງທໍາອິດ, ໄດ້ໃຫ້ຄວາມເຂົ້າໃຈກ່ຽວກັບວິວັດທະນາການ Fig-wasp ໃນລະດັບ genomic.
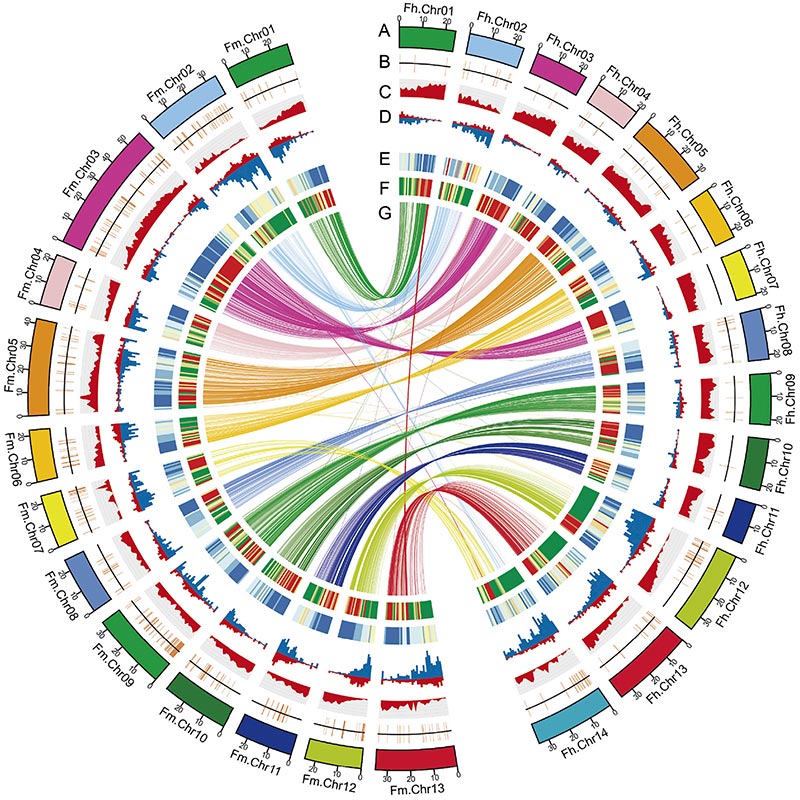 ແຜນວາດ Circos ກ່ຽວກັບລັກສະນະ genomic ຂອງສອງFicusgenomes, ລວມທັງ chromosomes, ການຊໍ້າຊ້ອນຂອງ segmental (SDs), transposons (LTR, TEs, DNA TEs), ການສະແດງອອກຂອງ gene ແລະ synteny | 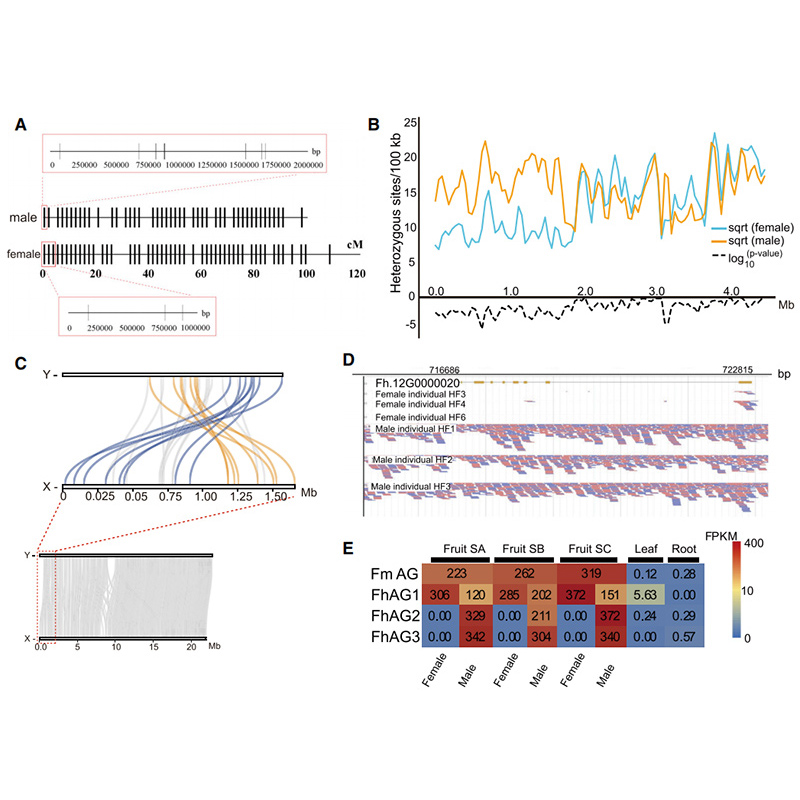 ການກໍານົດຂອງໂຄໂມໂຊມ Y ແລະການກໍານົດເພດຂອງຜູ້ສະຫມັກ |
Zhang, X. , et al."ພັນທຸກໍາຂອງຕົ້ນໄມ້ Banyan ແລະຕົວ pollinator Wasp ສະຫນອງຄວາມເຂົ້າໃຈກ່ຽວກັບ Coevolution Fig-Wasp."ຕາລາງ 183.4(2020).





