

Ensamblaje del genoma basado en Hi-C







Ventajas del servicio
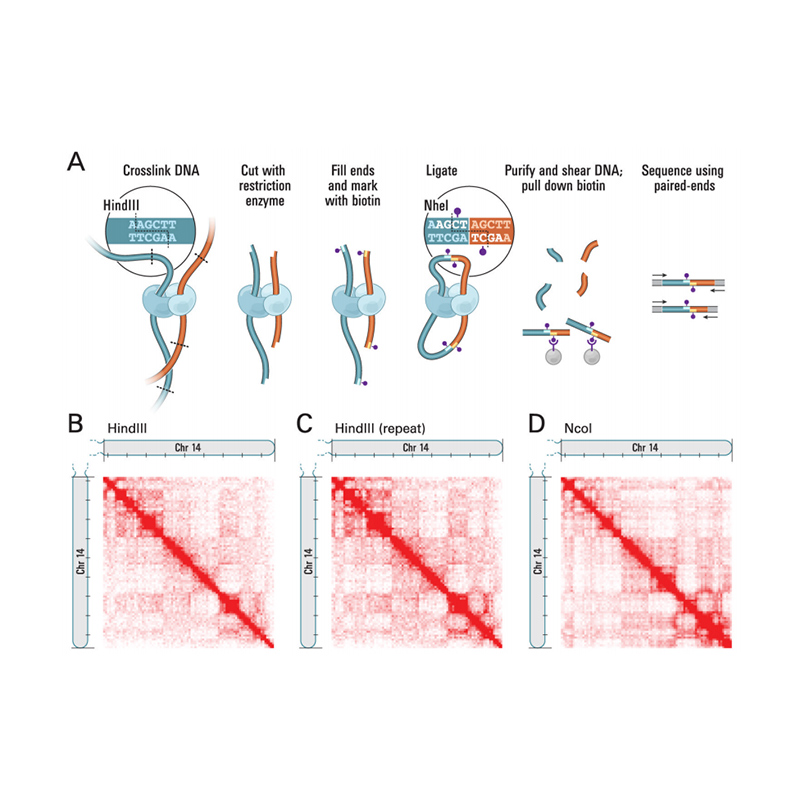
Descripción general de Hi-C
(Lieberman-Aiden E et al.,Ciencia, 2009)
● No es necesario construir una población genética para el anclaje contig;
● Una mayor densidad de marcadores conduce a una mayor proporción de anclaje de contigs, superior al 90%;
● Permite la evaluación y correcciones de conjuntos de genomas existentes;
● Tiempo de respuesta más corto con mayor precisión en el ensamblaje del genoma;
● Abundante experiencia con más de 1.000 bibliotecas Hi-C construidas para más de 500 especies;
● Más de 100 casos exitosos con un factor de impacto publicado acumulativo de más de 760;
● Ensamblaje del genoma poliploide basado en Hi-C; en el proyecto anterior se logró una tasa de anclaje del 100 %;
● Patentes internas y derechos de autor de software para experimentos de Hi-C y análisis de datos;
● Software de ajuste de datos visualizados de desarrollo propio que permite mover, revertir, revocar y rehacer bloques manualmente.
Especificaciones de servicio
|
Tipo de biblioteca
|
Plataforma | Longitud de lectura | Recomendar estrategia |
| Hola-C | Illumina NovaSeq | PE150 | ≥ 100X |
Análisis bioinformáticos.
● Control de calidad de los datos sin procesar
● Control de calidad de la biblioteca Hi-C
● Ensamblaje del genoma basado en Hi-C
● Evaluación posterior al montaje
Requisitos de muestra y entrega
Requisitos de muestra:
| Animal | Hongo | Plantas
|
| Tejido congelado: 1-2 g por biblioteca Celdas: 1x 10^7 celdas por biblioteca | Tejido congelado: 1 g por biblioteca | Tejido congelado: 1-2 g por biblioteca
|
| *Recomendamos encarecidamente enviar al menos 2 alícuotas (de 1 g cada una) para el experimento Hi-C. | ||
Entrega de muestra recomendada
Envase: tubo de centrífuga de 2 ml (no se recomienda papel de aluminio)
Para la mayoría de las muestras, recomendamos no conservarlas en etanol.
Etiquetado de muestras: las muestras deben estar claramente etiquetadas y ser idénticas al formulario de información de muestra enviado.
Envío: Hielo seco: las muestras deben empaquetarse primero en bolsas y enterrarse en hielo seco.
Flujo de trabajo del servicio

Diseño de experimentos

Entrega de muestra

Extracción de ADN

construcción de biblioteca

Secuenciación

Análisis de los datos

Servicios postventa
*Los resultados de demostración que se muestran aquí provienen todos de genomas publicados con Biomarker Technologies.
1.Mapa de calor de interacción Hi-C deCamptoteca acuminatagenoma.Como se muestra en el mapa, la intensidad de las interacciones se correlaciona negativamente con la distancia lineal, lo que indica un ensamblaje a nivel de cromosomas de alta precisión.(Relación de anclaje: 96,03%)
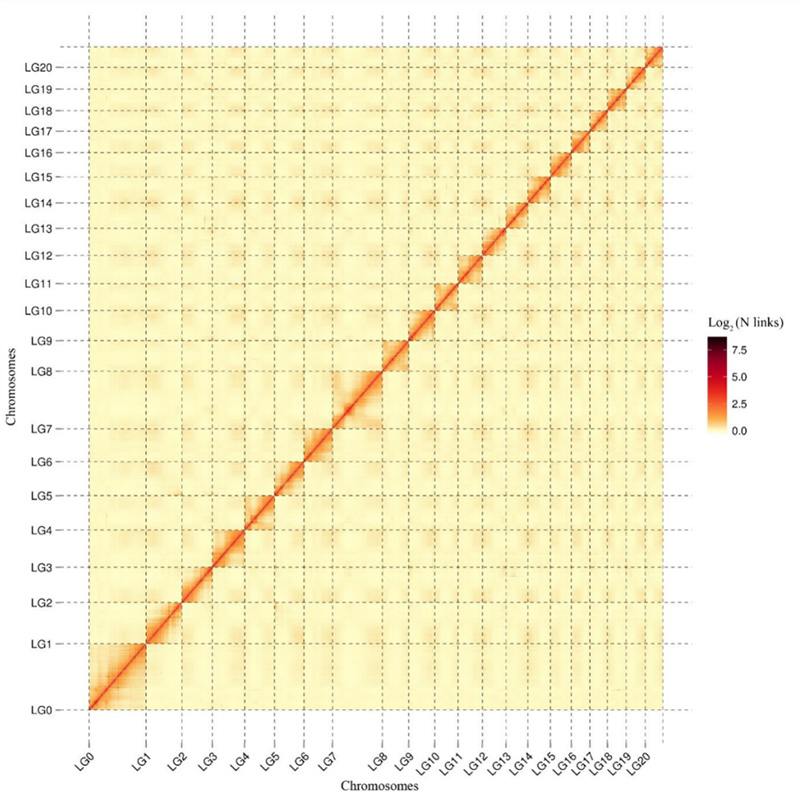
Kang M et al.,Comunicaciones de la naturaleza, 2021
2.Hi-C facilitó la validación de inversiones entreGossypium hirsutumL.TM-1 A06 yG. arboreumChr06
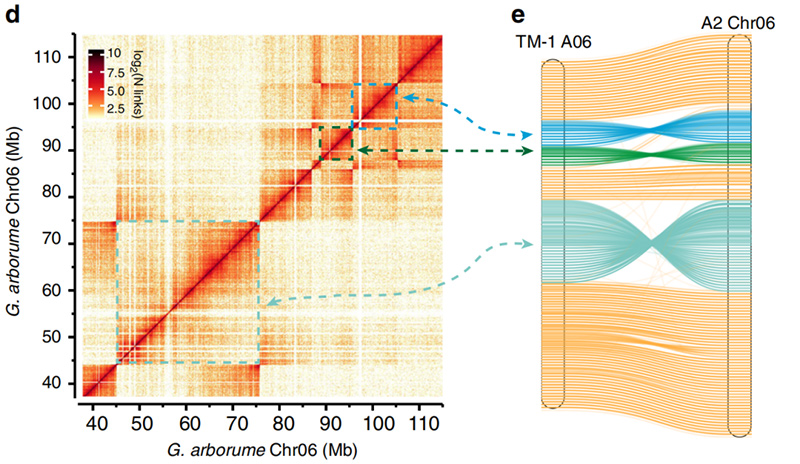
Yang Z y otros,Comunicaciones de la naturaleza, 2019
3.Ensamblaje y diferenciación bialélica del genoma de yuca SC205.El mapa de calor Hi-C muestra una clara división en cromosomas homólogos.
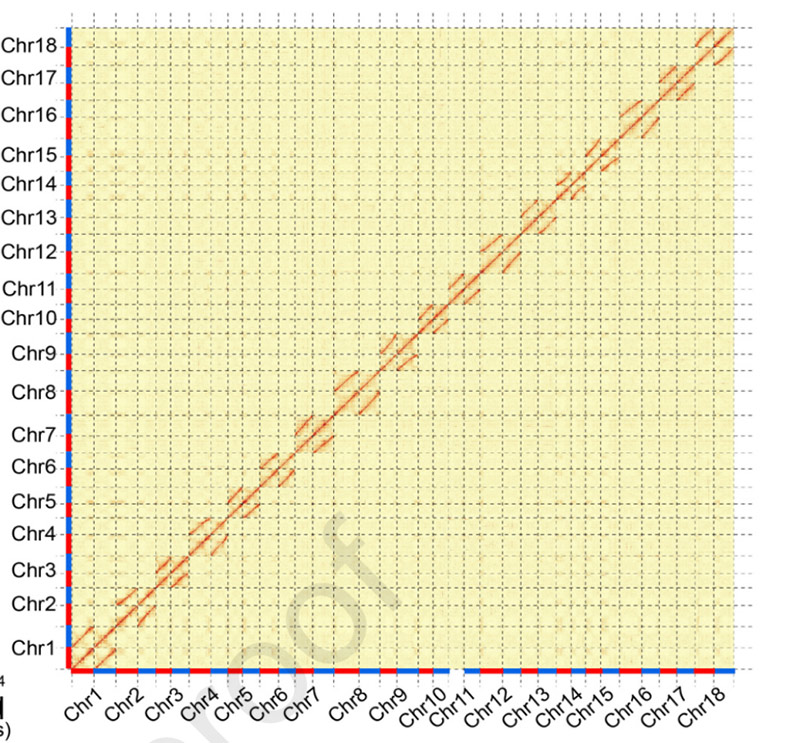
Hu W et al.,Planta molecular, 2021
4.Mapa de calor Hi-C en el ensamblaje del genoma de dos especies de Ficus:F.microcarpa(relación de anclaje: 99,3%) yF.hispida (ratio de anclaje: 99,7%)
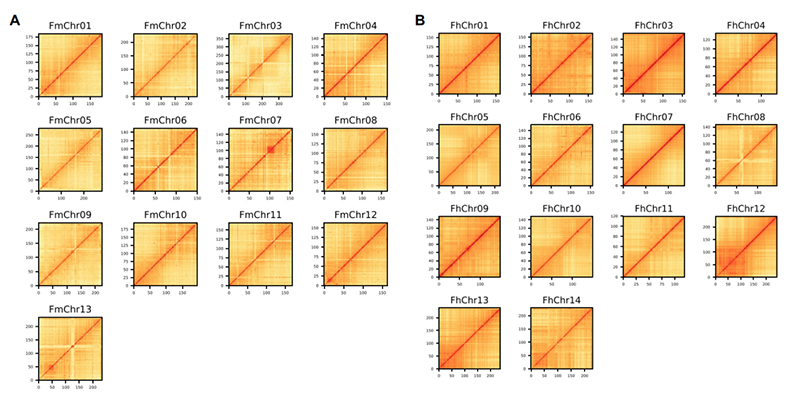
Zhang X y otros,Celúla, 2020
Caso BMK
Los genomas del baniano y la avispa polinizadora proporcionan información sobre la coevolución de la avispa y el higo
Publicado: Celúla, 2020
Estrategia de secuenciación:
F. microcarpa genoma: Aprox.84 X PacBio RSII (36,87 Gb) + Hi-C (44 Gb)
F. hispidagenoma: Aprox.97 X PacBio RSII (36,12 Gb) + Hi-C (60 Gb)
Eupristina verticillatagenoma: Aprox.170 X PacBio RSII (65 Gb)
Resultados clave
1. Se construyeron dos genomas de árbol de higuera y un genoma de avispa polinizadora utilizando secuenciación PacBio, Hi-C y mapa de ligamiento.
(1)F. microcarpaGenoma: se estableció un ensamblaje de 426 Mb (97,7% del tamaño estimado del genoma) con un cóntig N50 de 908 Kb, puntuación BUSCO de 95,6%.En total, Hi-C ancló secuencias de 423 Mb a 13 cromosomas.La anotación del genoma arrojó 29.416 genes codificadores de proteínas.
(2)F. HíspidaGenoma: Se produjo un conjunto de 360 Mb (97,3% del tamaño estimado del genoma) con un contig N50 de 492 Kb y una puntuación BUSCO del 97,4%.Un total de secuencias de 359 Mb fueron ancladas en 14 cromosomas por Hi-C y altamente idénticas al mapa de ligamiento de alta densidad.
(3)Eupristina verticillataGenoma: se estableció un conjunto de 387 Mb (tamaño estimado del genoma: 382 Mb) con un contig N50 de 3,1 Mb y una puntuación BUSCO del 97,7%.
2. El análisis genómico comparativo reveló una gran cantidad de variaciones estructurales entre dosFicusgenomas, que proporcionaron un recurso genético invaluable para los estudios de evolución adaptativa.Este estudio, por primera vez, proporcionó información sobre la coevolución de la avispa higuera a nivel genómico.
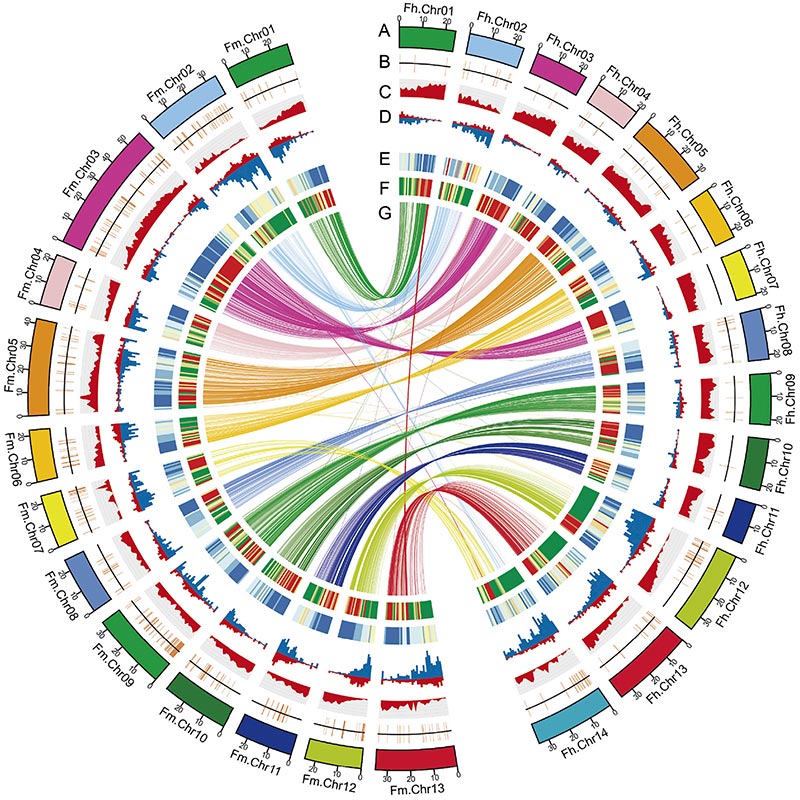 Diagrama circos sobre características genómicas de dos.FicusGenomas, incluidos cromosomas, duplicaciones segmentarias (SD), transposones (LTR, TE, ADN TE), expresión genética y sintenia. | 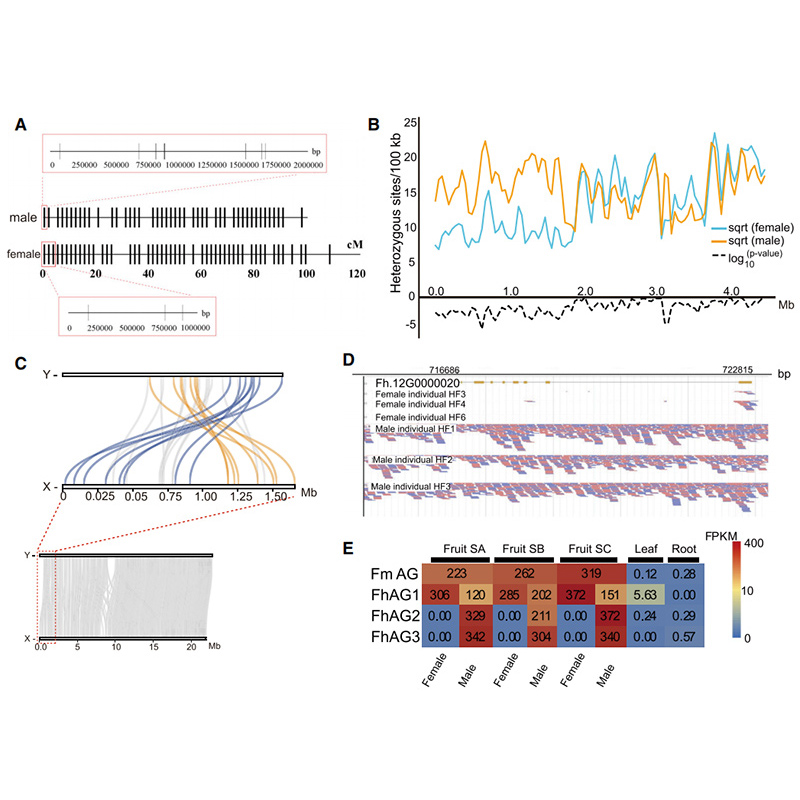 Identificación del cromosoma Y y gen candidato a la determinación del sexo. |
Zhang, X., et al."Los genomas del baniano y la avispa polinizadora proporcionan información sobre la coevolución higo-avispa".Celda 183.4(2020).





